Therapeutic interventions of acute and chronic liver disorders: A comprehensive review
2023-03-18FaresEMAliMostafaAbdElAzizElhamSharabAdelBakr
Fares EM Ali,Mostafa K Abd El-Aziz,Elham I Sharab,Adel G Bakr
Fares EM Ali,Adel G Bakr,Department of Pharmacology and Toxicology,Faculty of Pharmacy,Al-Azhar University,Assiut Branch,Assiut 71524,Egypt
Mostafa K Abd El-Aziz,Elham I Sharab,Faculty of Pharmacy,Al-Azhar University,Assiut Branch,Assiut 71524,Egypt
Abstract Liver disorders are one of the most common pathological problems worldwide.It affects more than 1.5 billion worldwide.Many types of hepatic cells have been reported to be involved in the initiation and propagation of both acute and chronic liver diseases,including hepatocytes,Kupffer cells,sinusoidal endothelial cells,and hepatic stellate cells (HSCs).In addition,oxidative stress,cytokines,fibrogenic factors,microRNAs,and autophagy are also involved.Understanding the molecular mechanisms of liver diseases leads to discovering new therapeutic interventions that can be used in clinics.Recently,antioxidant,anti-inflammatory,anti-HSCs therapy,gene therapy,cell therapy,gut microbiota,and nanoparticles have great potential for preventing and treating liver diseases.Here,we explored the recent possible molecular mechanisms involved in the pathogenesis of acute and chronic liver diseases.Besides,we overviewed the recent therapeutic interventions that targeted liver diseases and summarized the recent studies concerning liver disorders therapy.
Key Words: Liver disorders;Autophagy;Gene therapy;Anti-hepatic stellate cells;Cell therapy
INTRODUCTION
Chronic liver diseases are a significantly prevalent health problem contributing to the rising burden on countries daily.Specifically,liver cirrhosis - a result of chronic liver damage - is considered one of the well-known causes of morbidity and mortality all over the globe.According to Cheemerla and Balakrishnan[1],liver cirrhosis was responsible for the worldwide death of approximately 1.32 million patients in 2017.Not only that liver cirrhosis ranked 11thamong the leading causes of mortality,but it has also become a habitual cause of living with a disability[2].
Acute liver injury is characterized by an abrupt decline in hepatocyte function.Unlike liver cirrhosis,acute liver failure (ALF) typically has no underlying liver problem and worsens rapidly in days or weeks.Regarding etiology,hepatitis B viral infection and medication toxicity,particularly from acetaminophen (APAP),are the primary contributors to ALF.However,other types of hepatitis,autoimmune disorders,Wilson’s disease,and cardiovascular diseases are less common suspects for ALF[3].On the contrary,there are two classes of chronic liver injuries: Cholestatic conditions that block the bile flow and persistent hepatotoxicity.Various factors can lead to hepatotoxicity,such as hepatitis B viruses (HBV),hepatitis D viruses,and hepatitis C viruses (HCV),alcohol abuse,or non-alcoholic steatohepatitis (NASH).At the same time,biliary cholangitis,atresia of bile ducts,and primary sclerosing cholangitis can cause cholestatic injuries.Regardless of the causative agent,chronic hepatic inflammation causes liver fibrosis which,if not reversed,progresses to liver cirrhosis and hepatocellular carcinoma (HCC)[4,5].
Different physiological mechanisms have been involved in liver injury,including autophagy and their different types,microRNAs (miRNAs) and their crucial effect,inflammation,hepatic cell regulation role,and the main effects of transcription factors and inflammatory cytokines.Considering the therapeutic interventions for liver diseases,there are specific treatments that are basically dependent on the cause of the disease.For instance,alcohol cessation,acetylcysteine for APAP toxicity,antiviral medication for hepatitis viruses,and immunosuppressants for autoimmune hepatitis are considered[3].
Recent studies have discussed various interventions for liver disorders,such as antifibrotic agents,cell-based therapies,gut microbiota,different nanoparticle systems,gene therapy,and much more.Consequently,we aim to discuss the newly characterized pathophysiological mechanisms and the most appropriate and recent therapy discovered to be effective on acute and chronic liver disorders(Figure 1).
DIFFERENT PATHOPHYSIOLOGICAL MECHANISMS INVOLVED IN LIVER INJURY
Both initial liver damage and subsequent multiple organ failure (MOF) can be classified as parts of the pathophysiology of ALF.The mechanism of APAP-induced ALF is the most well-known in terms of the first liver injury.Glucuronidation and sulfation of APAP create harmless chemicals that are eliminated through the urine in nontoxic doses (4 g/d)[5].The residual APAP is transformed into the hazardous metabolite N-acetyl-p-benzoquinone (NAPQI) by cytochrome P450 enzymes (CYPs),which is then detoxified by bringing it to glutathione (GSH)[6].Interestingly,after overdosing on APAP,GSH is depleted after its conjugation with NAPQI,and the extra NAPQI binds to hepatocellular proteins causing mitochondrial oxidative stress and necrosis[7].NAPQI amount is enhanced during the decrease of GSH availability which will exacerbate the toxic effects of an APAP overdose.Antibiotics,antiepileptic medications,and ethanol activate CYPs and increase NAPQI production.Reduced GSH production is a result of fasting and malnutrition[6].
Moreover,the pathogenesis of secondary MOF appears to have several characteristics in common with severe sepsis.The innate immune response is triggered early in the course of a disease.It can be a response to heterotropic viruses’ pathogen-specific molecular patterns (PAMPs) or to damageassociated molecular patterns (DAMPs),which include histones,DNA,and high mobility group box molecules-1 proteins produced from wounded cells following hepatocyte apoptosis as a result of toxic causes[8].It is well known that the innate response involves a wide range of immune cells,such as monocytes,macrophages,dendritic cells,leukocytes,and natural killer cells.PAMPs and DAMPs are recognized by these cells,which then react and generate proinflammatory mediators like tumor necrosis factor α (TNF-α),interleukin (IL)-1,and IL-6,as well as reactive oxygen species (ROS),which trigger a systemic inflammatory response.
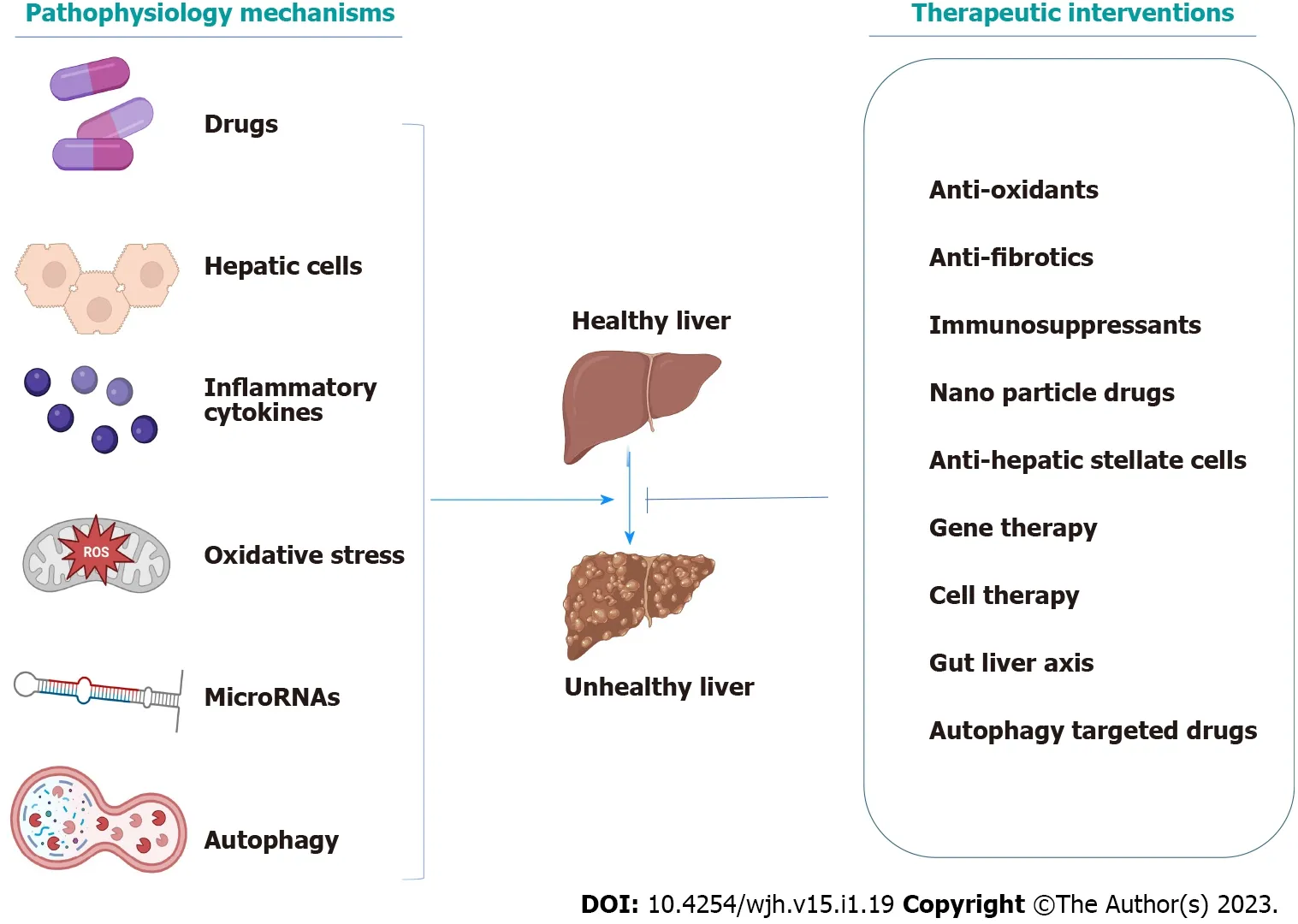
Figure 1 Graphical abstract.
Additionally,IL-17 and IL-10 contribute to the overall inflammatory response[8].Afterward,MOF is produced,and liver damage is still being brought on by ROS and cytokines.Proinflammatory cytokines entice neutrophils and encourage extravasation into the parenchyma of the liver.They begin to emit ROS and proteases once they are inside the parenchyma,which causes hepatocyte destruction.Promoting neutrophil extravasation into the hepatic parenchyma is greatly aided by mediators released from dying or dead hepatocytes and CXC chemokines.By releasing reactive oxygen intermediates and proteases once they have reached the hepatic parenchyma,neutrophils cause intracellular hepatocyte stress and oncotic necrosis[9].The vasodilatation of the peripheral microcirculatory leads to inefficient pulmonary oxygen exchange,decreased peripheral tissue oxygen supply,and subsequently,lactic acidosis,which finally causes hypotension.The most severe effects are on cerebral and renovascular tone,which results in hemorrhage,cerebral hyper-perfusion,and functional renal failure[8].The most common pathological mechanisms of acute and chronic liver disease are summarized in Table 1.
ROLE OF DIFFERENT CELL TYPES IN LIVER DISEASES
The liver is composed of two types of cells;hepatocytes,known as parenchymal cells,which constitute most of the liver and non-parenchymal cells.Around 10% of the liver’s mass comprises nonparenchymal cells that include liver sinusoidal endothelial cells (SECs),hepatic stellate cells (HSCs),biliary cells,Kupffer cells (KCs),and immune cells such as neutrophils,natural killer cells,and infiltrating macrophages[10].Whenever the liver is exposed to a harmful substance,both parenchymal and non-parenchymal hepatic cells take a role in the onset of liver fibrosis and cirrhosis.
Understanding the etiology of chronic liver disorders is essential for their prevention,slowing their progress,and advancing different treatment options.There are various etiologies to chronic liver disease,from alcoholic liver disease (ALD),non-alcoholic fatty liver (NAFLD),steatohepatitis,and chronic viral hepatitis,to other genetic,autoimmune,drugs,or cryptogenic liver diseases.Among the different etiologies of liver disorders,alcohol abuse is the most common cause.As a result of excessive alcohol consumption,the condition of alcoholic liver worsens to fatty liver and chronic steatohepatitis,which in turn triggers liver fibrosis,cirrhosis,or even HCC[11].The non-ALD shares the same fate as the ALD but is correlated with metabolic syndrome[12].Also,different types of hepatitis viruses can result in chronic liver disease,especially hepatitis B and C;hence,they are considered a major concern for cirrhosis and liver cancer[13].Concerning the less common causes of liver disorders,genetic factors such as hemochromatosis,alpha-1 antitrypsin deficiency,Wilson’s disease,and autoimmune hepatitis can all contribute to irreversible cirrhosis[14].Additionally,hepatotoxic drugs,primarily APAP,followed by idiosyncratic drugs inducing liver injuries,such as antibiotics,nonsteroidal anti-inflammatory drugs,herbal remedies,and statins,can cause the liver to progress to liver fibrosis and cirrhosis[15].When the liver is exposed to any of the above-mentioned destructive agents,liver cells undergo a remodeling process to compensate for the damage.
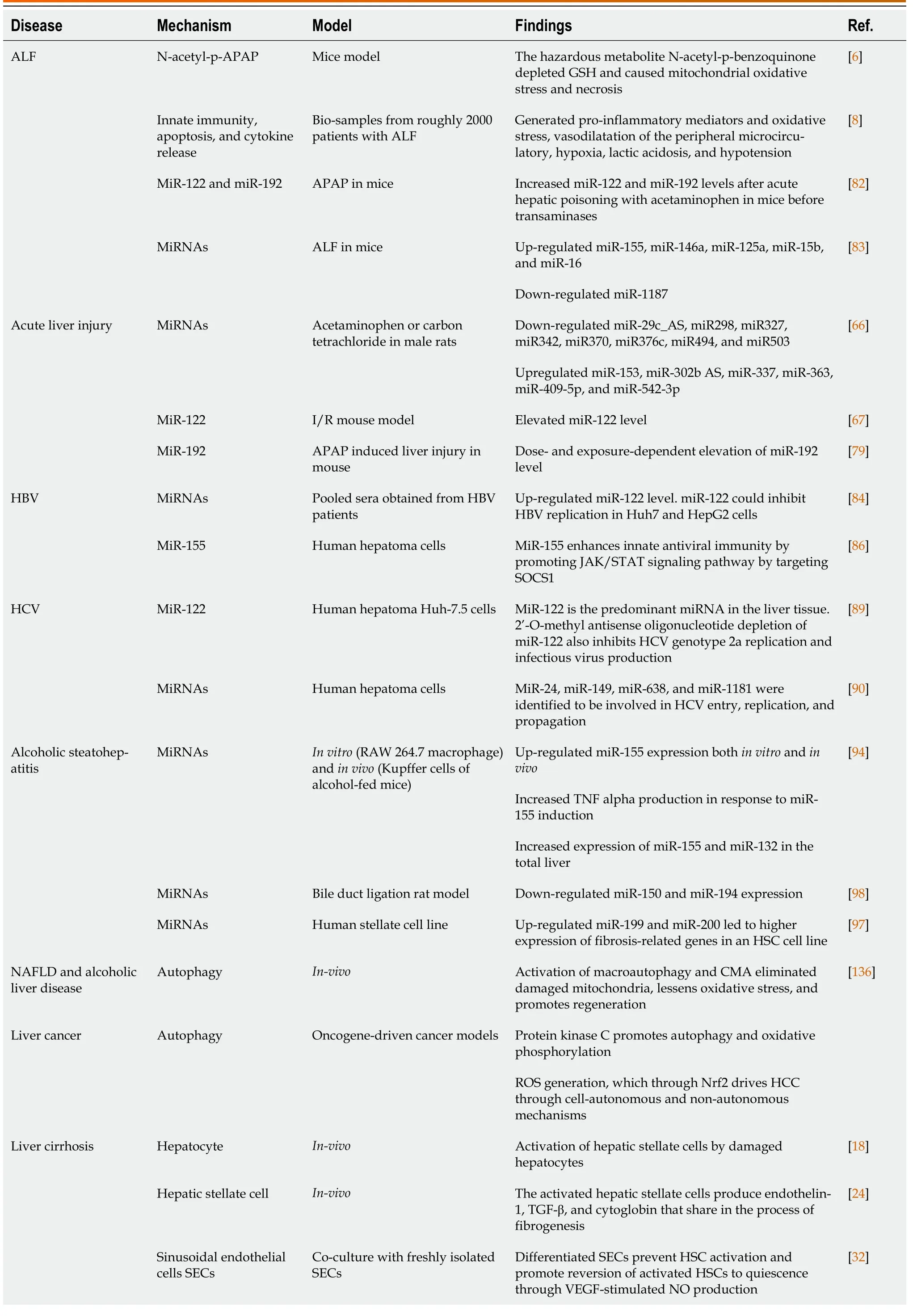
Table 1 Summarized the common pathological mechanisms of acute and chronic liver diseases

ALF: Acute liver failure;APAP: Aminophenol;HBV: Hepatitis B virus;HCV: Hepatitis C virus;SECs: Sinusoidal endothelial cells;VEGF: Vascular endothelial growth factor;HSCs: Hepatic stellate cells;TGF-β: Tumor growth factor-beta;CMA: Chaperone-mediated autophagy;HCC: Hepatocellular carcinoma;Nrf2: Nuclear factor erythroid 2-related factor 2;NAFLD: Non-alcoholic fatty liver;TNF: Tumor necrosis factor;GSH: Glutathione;ROS:Reactive oxygen species;miRNA: MicroRNA.
Hepatocytes play a complex role in the progression of cirrhosis since they are the main constituent of the liver and are particularly susceptible to harm from hepatotoxic substances[16].Hepatocytes produce most of the matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases,which regulate the extracellular matrix deposition and thus participate in the process of liver cirrhosis[17].Damaged hepatocytes activate HSCs,increase the ability of myofibroblasts to synthesize fibrous tissue,and produce ROS and other fibrogenic mediators[18].The persistence of fibrosis induces hepatocytes to become hypoxic and produce large amounts of tumor growth factor-beta (TGF-β),a powerful stimulator of fibrogenesis[19].Additionally,recent studies showed that hepatocyte telomere shortening and aging is a possible factor contributing to fibrosis and is thus implicated in the pathogenesis of cirrhosis[20].
HSCs are primarily in charge of regulating and storing vitamin A or retinol,and they are found in the subendothelial space between hepatocytes and SECs.ROS,cytokines,and growth factors,such as TNF-α and TGF-β,respectively,can activate these quiescent cells,causing them to synthesize a lot of extracellular matrixes,which can form a scar in the space of the disease[21-23].In addition,the activated HSCs produce endothelin-1,TGF-β,and cytoglobin that share in the process of fibrogenesis[24-26].However,recent studies showed that the delivery of berberine nanoparticles could inhibit the proliferation of HSCs and reverse the damage resulting from fibrosis[27].
SECs are an important type of liver cells,surrounded by the bloodstream from one side and hepatocytes from the other side[28].Morphologically,liver SECs are characterized by transcellular pores known as fenestrae,which are essential for transporting nutrients and other components from the blood to the hepatocytes and vice versa.Fenestrae is important for normal liver function and plays a great role in maintaining liver homeostasis and regeneration[29].In pathological conditions,SECs lose their fenestrae and become capillarized,impairing proper liver function[30].Furthermore,they encourage fibrogenesis by activating HSCs by releasing IL-33[31].In contrast,several studies have documented that differentiated liver SECs can encourage the reversion of activated HSCs to the quiescent form and thus stop the progress of fibrosisviamodulating vascular endothelial growth factor (VEGF)-stimulated NO release[32].
KCs are liver macrophage cells that comprise an average of 85% of body macrophages and are present in hepatic sinusoids.KCs are necessary for innate and adaptive immunity as they deal with detrimental pathogens entering the liver from the portal vein[33].As a result of liver injury,KCs get activated and respond by producing various cytokines,ILs,and chemokines[16].Additionally,NO produced by KCs,together with TNF-α,TGF-β,and platelet-derived growth factors (PDGFs),activate HSCs,causing an excess of extracellular matrix to be produced[34].Although KCs produce death ligands and contribute to liver fibrogenesis and fibrosis[35],they are not a suitable target for therapeutic interventions due to their crucial host defense function.
ROLE OF CYTOKINES,TRANSCRIPTION FACTORS,AND ROS IN HEPATIC INJURY
Cytokines are bioactive molecules made by several types of liver cells that are essential in the progression of liver cirrhosis[36].They consist of TNF-α,PDGF,interferons (IFNs),ILs,TGF-β,chemokines,and adipokines.Several important biological processes,such as hematopoiesis,immunology,inflammation,and body development,are mediated by cytokines.However,they are also linked to several illnesses,including liver disorders,rheumatoid arthritis,and atherosclerosis[37].A significant coordinated program of cellular and molecular alterations in liver cirrhosis results in a potent fibrotic response.Cytokines are involved in the combative signaling pathways that regulate the activation of HSCs and fibrogenesis[38].
PDGF is the most powerful HSCs activator concerning all polypeptide growth factors.According to the degree of fibrosis,it seems to be overexpressed,enhancing its receptors and their activity in fibrous tissue[39].Mainly in reaction to diverse stimuli,including viruses,chemicals,or mechanical injury,KCs manufacture and release PDGF[40].When PDGF is released,it attaches to a particular receptor on the HSCs’ membrane,activating transcription factors and matching signal molecules involved in the process[41].This causes the activation of its target genes,which are downstream of the receptor,as well as the activation of HSCs.It has been shown that PDGF (P38-MAPK) increases the activity of C-Jun Nterminal kinase,extracellular signal-regulated kinase (ERK) 1/2,MMP,TIMP,protein kinase B/AKT pathways,and P38 mitogen-activated protein kinase[39].
Transforming growth factor-beta is the strongest known fibrogenic inducer during liver cirrhosis[42].It is released by all types of hepatic cells in response to unpleasant stimuli and is essential for developing and spreading cirrhosis and liver fibrosis.In fibrotic diseases,TGF-β is abundantly expressed and reaches its peak in cirrhosis[43].TGF-β pro-fibrogenic impact is carried out by boosting the production of HSCs and ECM while inhibiting MMPs,which results in an excessive buildup of collagen fibers and aids in the progression of liver fibrosis[44].Additionally,it has been demonstrated that TGF-β causes hepatocyte death and inhibits DNA synthesis[38].
TNF-α is a pro-inflammatory cytokine generated during inflammation and oversees various cell signaling processes.HSCs,KCs,monocytes,and macrophages secrete it[45].According to a study showing that TNF-α is a mediator of hepatotoxicity and inflammation in many liver diseases,hepatocellular injury followed by inflammation and activation of the innate immune system leads to earlystage liver fibrosis,which in turn causes HSC activation and ECM deposition[46].In addition,TNF-α contributes to ECM deposition by enhancing the expression of TIMP-1 in HSCs[47].TNF-α has complex and sometimes conflicting effects on HSCs and fibrosis.TNF-α,on the other hand,has also been demonstrated to have an anti-fibrogenic impact in rat’s HSCs by lowering GSH and decreasing procollagen 1 expression.TNF’s function in fibrogenesis is debatable,and it is unknown exactly how TNF receptors contribute to the activation of HSCs.Researchers demonstrate that loss of both TNF receptors decreased pro-collagen 1 expression,slowed HSC proliferation,and impaired PDGF-induced promitogenic signaling in HSC from wild-type,TNF-receptor-1 (TNFR1) knockout,TNFR2 knockout,or TNFR1/R2 double knockout (TNFR-DKO) mice.In response to PDGF,TNFR-DKO HSC showed decreased AKT phosphorylation andin vitroproliferation.However,these effects were not replicated in TNFR2 knockout HSC.Additionally,in primary mouse HSC,TNF binding to TNFR1 was necessary for MMP-9 expression.Neutralizing antibodies against TNFR1 and TNFR2 confirmed these findings in the human HSC cell line LX2.Additionally,compared to wild-type or TNFR2 knockout mice,TNFR-DKO and TNFR1 knockout animals showed lessin vivoliver damage and fibrogenesis after bile duct ligation(BDL)[48].
Oxidative stress is frequently described as a general imbalance between oxidizing and reducing substances in the cell.The signaling transduction pathways are governed by these redox states.Numerous human disorders,particularly chronic liver diseases,have been linked to the development of ROS[49].The production of ROS is crucial in causing liver injury and kicking off hepatic fibrogenesis.Oxidative stress alters lipids,proteins,and DNA,causing hepatocytes to necrotize and apoptosis and escalating the inflammatory response[50].
Additionally,ROS directly activates HSCs and encourages the synthesis of profibrogenic mediators from KCs and circulating inflammatory cells,which leads to the beginning of fibrosis[51].Regardless of their underlying causes,almost all liver illnesses have been found to exhibit oxidative stress[52].Prooxidants are ROS that can harm liver cells and whose levels may be raised by some medications,infections,environmental exposures,tissue damage,and other factors.Oxidative stress can be caused by increased prooxidant production,a reduction in antioxidant levels,or a shortage of antioxidants.Signaling,regulation,and redox balance of the liver system are biased by molecular redox switches,oxygen detection by the thiol redox proteome,NAD/NADP,and phosphorylation/dephosphorylation systems.ROS rapidly interact with all biological macromolecules due to their reactivity.The phosphodiester bonds that keep the bases in RNA and DNA together are cleaved by ROS,causing RNA and DNA to lose their chain structure.In a process known as lipid peroxidation,polyunsaturated fatty acids are another important target for oxidation by ROS.This process disturbs the normal structure of the membrane and results in necrosis.Additionally,since cysteine is necessary for the function of enzymes,ROS,particularly the hydroxyl radical,oxidizes cysteine residues in proteins to form disulfides,sulfoxides,or sulfonic acids.Additionally,oxidative stress promotes fibrogenesis by raising toxic cytokines such as TNF-α,IL-6,and TGF-β[53].
ROS generated by the NADP/NADPH oxidase system can control the cellular redox environment in hepatocytes and KCs.NADPH oxidase activation is the main ROS source in myofibroblasts and the stimulation of profibrogenic pathways[54].It is regarded as the main producer of superoxide anion and hydrogen peroxide,the two most damaging ROS contributing to liver damage from oxidative stress[55].NADPH oxidase inhibition is emerging as a target for antifibrotic treatment since NADPH oxidase activation may constitute a central mechanism in fibrosis[56].
The activities of various antioxidant enzymes,whose expression is controlled by several redoxsensitive transcription factors like nuclear factor kappa-light-chain-enhancer of activated B cells [nuclear factor-kappaB (NF-κB)] and nuclear factor erythroid 2-related factor 2 (Nrf2),may have an impact on the generation of ROS[57].Quiescent HSCs lack NF-κB in contrast to activated HSCs,which suggests that a redox-sensitive activation of NF-B might govern the expression of NF-B-targeted genes and provide a suitable cellular redox threshold for quiescent HSCs to enter the proliferative cycle[58].In support of this theory,it has been shown that blocking NF-κB activity shields rats from the onset of hepatic fibrosis.The suppression of Nrf-2 may also change the expression of antioxidant enzymes,disrupting the cellular redox environment and impacting HSC proliferation,cell death,and collagen formation,all of which contribute to liver fibrosis[49].
Additionally,ROS-sensitive cytokines help activate HSCs during inflammation by receiving paracrine cues from immune cells.Hepatic fibrosis progresses more quickly due to the activated HSCs’increased receptivity to PDGF and TGF-β[40].TGF-β boosts the generation of ROS while lowering the level of reduced GSH.The production of the collagen I protein is increased when lipid peroxidation is increased,and anti-oxidant defenses like GSH,catalase,or superoxide dismutase are decreased[59].
ROLE OF MIRNAS IN HEPATIC DISEASES
MiRNAs are a group of tiny,non-coding endogenous RNA molecules with a high degree of chemical stability (22 nucleotides).MiRNAs have been thoroughly investigated since their discovery in 1993[60]because of their function in RNA-induced posttranscriptional gene silencing.One of the most prevalent adult hepatic miRNAs,miR-122,controls several important gene networks,including lipid metabolism,cell differentiation,and the hepatic circadian rhythm[61].Recently,miR-223 is thought to interfere with the development and homeostasis of the immune system as well as it has an important role in inflammatory disorders and other liver disorders[62].Moreover,MiR-223 also controls the nucleotide-binding oligomerization domain-like receptor (NLR) inflammasome by targeting the NLR protein 3 (NLRP3) 3′-untranslated regions[63].Notably,different cell types require NLRP3 inflammasome to start the inflammatory reaction and the production of ILs.Accordingly,overexpression of miR-223 reduces IL-1 production from the inflammasome and prevents NLRP3 protein formation.Additionally,miR-223 may prevent macrophage hyperactivation[64].
Recent evidence showed that numerous liver disorders,including viral hepatitis,alcohol-induced liver damage,drug-induced liver injury,NAFLD,cirrhosis,and HCC,have dysregulated the expression of the miR-223 gene.Markedly,Weseslindtneret al[65] revealed that,the elevation of miR-106a,miR-122,and miR-197 levels in patients with severe acute viral hepatitis.Interestingly,Fukushimaet al[66]made a thorough comparative microarray study and looked at how different miRNAs changed in rats after receiving APAP and CCL4 and discovered that eight miRNAs were downregulated while six miRNAs (miR-153,miR-337,miR-363,miR-302b AS,miR-409-5p,and miR-542-3p) were upregulated in both hepatotoxicity models.
Since miR-122 is very liver-specific and makes up around three-quarters of the entire miRNAs that the liver expresses,it has been extensively studied concerning liver damage[67-69].It is highly expressed in hepatocytes because of liver-specific transcriptional regulation under the effect of hepatic transcription factors[70].Further,it seems to be elevated in the majority of liver disorders,including HCV and HBV,in addition to ALD,drug-induced liver damage,NAFLD,and HCC[71-74].Along with this,loss of miR-122 is seen during hepatocellular carcinogenesis due to hepatic cell dedifferentiation[75,76].In acute and chronic liver disorders,miR-122 serum/plasma levels are correlated to hepatic necroinflammation,elevated aminotransferase levels,liver injury,and cell death[77].
Not only miR-122 but miR-192 as well was elevated in the mice sera after APAP administration compared to controls in a dose-dependent and exposure-dependent manner.In this context,the levels of those miRNAs were enhanced sooner than the levels of serum transferases[78],highlighting that they can be used diagnostically superior to the conventional ALF indicators[79].Similarly,the serum levels of miR-122 were also elevated in the I/R mice models,and they were connected to both aspartate aminotransferase (AST)/alanine aminotransferase (ALT) levels and the hepatic cell death identified by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling.Asin vitrostudies showed that miR-122 levels increase in the supernatant after hepatocyte injury.The presence data imply that miR-122 may replace hepatocyte mortality in liver damage[67].Additionally,the elevation of miR-122 and miR-192 in the sera of patients with APAP-induced ALF could be confirmed,and these findings concur with results from high-throughput sequencing of patients who had taken too much APAP[69].Krauskopfet al[69] showed that,compared to controls,the serum levels of 36 types of miRNAs were higher in these individuals.Additionally,following APAP overdose,miR-122,miR-192,miR-194,miR-210,and miR-483 were shown to be reinforced in the liver.
In ALF,a considerable downregulation of miR-122 is seen in the injured liver in both acute and chronic liver injury,and it showed an inverse correlation between hepatic damage and ALT levels,suggesting that it may play a role in human ALF.When paraquat was administered to humans,miR-122 was noticeably upregulated,whereas miR-483 and miR-711 were concurrently downregulated.This is consistent with what was shown in the rats given an APAP overdose[78,80].Zhanget al[81] discovered that blood levels of miR-122 and miR-192 were increased after acute hepatic poisoning with APAP in mice before transaminases,particularly ALT,were raised.However,it was shown that the miRNA levels in liver tissue were lower.Since these miRNAs may be detected before the liver experiences apparent cell death,they may serve as a more accurate indicator of liver failure than liver enzymes[82].Recent studies showed that miR-15b,miR-16,miR-125a,miR-146a,and miR-155 were considerably upregulated during ALF in mice,while miR-1187 showed a significant down-regulation[83].
Hepatitis B e antigen (HBeAg) positive patients had much greater blood levels of miRs than those with HBeAg negative,especially miR-122 and miR-194,which showed the greatest differential expression[84].Additionally,it has been shown that the expression of miR-122,miR-638,miR-572,miR-575,miR-638,and miR-744 was dysregulated in chronic HBV patients;these miRs were significantly more abundant in HBV than AST or ALT.MiR-122,miR-572,miR-575,and miR-638 were more abundant than miR-744[85].In human hepatoma cells,HepG2,miR-155 has been shown to contribute to antiviral immunity against HBV infection[86].An initial therapeutic response to IFN (independent relationship with early virologic response) may be predicted in HBV patients using a miR profile of 11 miRs for example,hsa-let-7a,hsa-miR-30a,hsa-miR-106b,hsa-miR-198,hsa-miR-1224-5p,and hsa-miR-1290.It has been demonstrated that certain miRs might play a function in the HBV life cycle[87].According to studies,specific miRs have been shown to affect HCV infection or be affected by the virus.There is still much to learn about how miR-122 interacts with the HCV genome[88].
However,miR-122 expression is unaffected by viral infection or replication.Recently,Randallet al[89]looked at miR-21 and miR-122 expression in the liver biopsy samples from patients infected with HCV and controls.They established that miR-122 levels were inversely linked to the fibrotic stage,ALT,and AST but that miR-21 levels were positively linked.It was suggested that rather than levels of expression,fibrosis might be brought on by dysregulation of miR-21 and miR-122.MiRs 24,149,638,and 1182,among others,share in HCV entrance,replication,and spread[90].The tumor suppressor “deleted in liver cell-1” protein was shown to be highly dependent on miR-141 activation,miR-141-targeted downregulation,and depletion for sustained HCV propagation.According to research on the association between HCV and the levels of miR-29 in both HSC and hepatocytes,HSC stimulation results in miR-29 down-regulation[91].The overexpression of miR-29 in infected cells reduced HCV replication by 70% and inhibited the growth of HSCs and collagen synthesis.When comparing the livers of HCV with non-SVR,miR-29a,b,and c levels were higher[92],indicating a potential function for these biomarkers in monitoring the effectiveness of anti-HCV therapy.
In alcoholic steatohepatitis,miRs are crucial immune response regulators and activators of the innate immune system[93].Alcohol-induced gut leakiness,which permits endotoxin to enter the blood and begin liver damage,has been shown to play a critical role in ALD and to increase miR-122 expression.It is shown that inducing miR-155 and -132 causes KCs to release higher TNF-α in response to lipopolysaccharide (LPS)[94].Hepatic miRs 182,183,705,1224,and 199a-3p are modulated by endotoxemia and alcohol use directly[95].Alcohol specifically targets and upregulates the miR-155 gene in macrophages,which controls the production of TNF-α[96].Prolonged alcohol exposure also stimulates the miR-155 gene in KCs and RAW264.7 macrophages.As a result,miR-155 upregulation might be engaged in the oxidative stress and LPS pathways,thus promoting the development of ALD[94].
There is mounting evidence that miRs,namelyviacontrolling gene expression in HSCs,are important regulators of hepatic fibrogenesis.The advancement of liver fibrosis has been linked to the miR-199 and miR-200 family’s expression.Patients with fibrotic livers had higher levels of the miR-199 and miR-200 families,and upregulation of these miRs led to considerably higher levels of the genes related to fibrosis in a cell line of HSC.In a fibrosis model of BDL in rats,miR-150 and miR-194 levels were significantly lower than in animals with a sham procedure.Furthermore,in a human stellate cell line called LX2,it has been shown that overexpressing miR-150 or miR-194 through the reduction of c-myb and rac1 expression can reverse the activated stellate cells (i.e.,expression of collagen and alpha-smooth muscle actin genes).Therefore,miR-150 and miR-194 may represent promising therapeutic targets for fibrosis treatment[97,98].
ROLE OF AUTOPHAGY IN LIVER DISEASES
Autophagy is a self-eating catabolic mechanism in eukaryotic cells that ends in the lysosome[99,100].In addition to its anti-aging function,autophagy plays a significant role in immune response and organ homeostasis[101,102].Numerous pathological disorders,such as obesity and type 2 diabetes,inflammatory and viral diseases,neurodegenerative diseases,and cancer,exhibit autophagy dysregulation[103,104].There are distinct phases of autophagy;induction,phagophore development,autophagosome creation,autolysosome formation,and destruction[105,106].Atg molecules participate in several complexes crucial for triggering autophagy and creating autophagosomes[107].The unc-51-like kinase 1 complex (Atg1 in yeast) is activated first,then beclin 1 (Atg6 in yeast),and followed by a series of Atg proteins that result in the production of autophagosomes,with LC3 (Atg8 in yeast),being one of them[108].Further processing of LC3 results in the formation of LC3-I and LC3-II[109].As soon as the autophagosome gets created,a blockade of autophagic flux at later stages will suppress the autophagosome’s ability to be cleared,ultimately leading to autophagy-dependent cell death[110].To date,macroautophagy,microautophagy,and chaperone-mediated autophagy (CMA) are the three main types of autophagy that have been characterized[111,112].
AUTOPHAGY AND THE IMMUNE SYSTEM
Lately,researchers have investigated the relationship between autophagy and the immune system[113,114].There have been documented non-canonical macroautophagic processes that create lysosomefusing autophagosomes[115].Only a portion of the Atgs equipment is utilized.Due to its significance in immunological modulation,LC3-associated phagocytosis (LAP) has received the most attention[116,117].LAP draws LC3-II to the phagosomal membraneviainnate immune receptors,such as toll-like receptors,where macrophages consume it.The crucial part that CMA plays is antigen presentation and aging,which has also garnered attention[118].Innate immunity’s ability to hinder macrophage autophagy is also associated with autophagy.Innate immunity and autophagy interact because IFN-α stimulates autophagy in macrophages[119].
AUTOPHAGY AND CELL DEATH
In some circumstances,autophagy can either serve as a defense mechanism or contribute to cellular death[120,121].The main way autophagy contributes to cellular death is through its influence on apoptosis.Apoptosis and autophagy are linked,and these two cellular destructing processes influence one another[122,123].This is crucial in the demise of liver cells[124].Autophagy generally prevents caspase-dependent apoptosis from being induced,whereas apoptosis-related caspase activation halts the autophagic process.
Along with these results,Niet al[125] documented that necrosis and necroptosis are caused by caspase-independent cell death,which is closely linked to autophagy.Cells harmed by the tumor suppressor gene p53 are removed by the induction of apoptosis[126].In addition to being engaged in autophagy,the mechanistic target of rapamycin (mTOR)/AKT pathway also inhibits apoptosis.For the destiny of damaged cells,p53 and AKT/mTOR must coexist in equilibrium[127].Numerous proteins linked to autophagy,including Atgs and BECN1,also played a role in ferroptosis.Additionally,erastin,an activator of ferroptosis,caused the formation of autophagosomes,and activation of autophagy resulted in ferroptotic cell death,maybe because of the ferritin being broken down by ferritinophagy[128].
Autophagy and inflammation
Autophagy and the liver’s inflammatory response are tightly related.The same inhibitory mechanisms govern autophagy and inflammasome but are regulated by various input pathways.Procaspase-1 activation results from the activation of the NLRP3 inflammasome,which is often triggered by pathogen- or danger-associated molecular patterns[129],which will further stimulate the synthesis of IL-1 and IL-18 that causes pyroptotic cell death.Moreover,the activation of autophagy by caspase-1 prevents these occurrences.Additionally,autophagy decreases inflammasome activation by destroying inflammasomes in autophagosomes and removing damaged cytoplasmic organelles that,in the absence of autophagy,would otherwise create DAMPS and increase inflammasome activation[130].On the other hand,when autophagy is diminished,the pro-inflammatory IL-1 is produced more often due to the negative association between inflammasomes and autophagy[131,132].Although the connection between NLRP3 and autophagy is not entirely understood,recent research has indicated that NF-κB activation can similarly modify NLRP3 and autophagy[133].
Given the preceding,it is not surprising that many autophagy reviews emphasize the contrasting impacts that autophagy may have on the same biological process by using the phrase “double-edged sword”[134].Cancer[135] and viral infections[101] are prominent fundamental paradigms.The fact that autophagy exhibits Jekyll-like and Hyde-like characteristics depending on the cells involved is another trait exclusive to the liver.Hepatocytes in NAFLD and ALD exhibit protective macroautophagy and CMA (in NAFLD).It eliminates damaged mitochondria,lessens oxidative stress,and promotes regeneration.In macrophages,macroautophagy reduces liver fibrosis and inflammation while promoting fibrosis-activated stellate cells.It is preventative in the early stages of HCC but might be damaging in the later stages[136].Both the non-parenchymal sinusoidal cells of the liver and the hepatocytes depend on autophagy for proper liver function[137],and autophagy abnormalities are linked to most liver illnesses’ pathogenesis[138].Autophagy disorders are linked to both common conditions like alcoholic and NAFLD or viral hepatitis and uncommon conditions like Wilson disease and alpha 1 antitrypsin deficiency[139,140].Due to the 6-12 mo half-life of hepatocytes,impaired autophagy also contributes to the accumulation of toxic hepatocyte byproducts.A large number of xenobiotics must also be processed by the liver,and autophagy is a cytoprotective mechanism[141].
Therapeutic interventions for acute and chronic liver diseases
Cirrhosis of the liver,the end stage of liver fibrosis after chronic liver damage,used to be cured by nearly liver transplantation only.That is why researchers used to focus on preventing liver cirrhosis by eradicating the cause and reversion of fibrosis.However,if liver cirrhosis develops,treatment is restricted to preventing the progression of the complications and avoiding the need for liver transplantation[142-144].Besides removing the cause,various categories of treatments have proven to be beneficial in preventing fibrosis progression or regression,such as antioxidants,and antifibrotic agents,including phyto drugs[144-147].Viaunderstanding the process of fibrogenesis,various mechanisms implicated in this process would be potential for the reversion of fibrosis and cirrhosis.Here in,we discuss several conventional and novel therapeutic interventions that showcased,by recent data,the ability to modulate liver fibrosis and cirrhosis.The recent therapeutic interventions are summarized in Table 2.
Antioxidants
Oxidative stress is well known to play a detrimental role in developing liver cirrhosis.When ROS production exceeds antioxidants level,cellular signaling pathways alterations eventually result in liver damage[148].For this reason,antioxidants received much attention and extensive study to prevent and treat various liver disorders.Silymarin is an herbal extract that consists mainly of silybin,which is responsible for the activity of silymarin.Free radical scavenging activity and inhibition of lipid peroxidation have been exhibited as reasons for the antioxidant activity of silybin[149].Selenium is an essential element for the GSH antioxidant system in our bodies that has been extensively studied for its antioxidant activity in various cases of liver damage[150].Selenium showed the ability to decrease DNA damage and hepatocyte necrosis against cyclophosphamide-induced oxidative stress[151].In cadmiuminduced acute liver injury,selenium nanoparticles decreased liver toxicity by boosting the Nrf2 pathway[152].In chronic liver injury,selenium is reported to mitigate lipid peroxidation and decrease other oxidative stress biomarkers,especially when combined with the natural antioxidant gum arabic[153].A study investigating the effect of curcumin,selenium,and silymarin showed that the combination of selenium,curcumin,and silymarin ameliorates the oxidant/antioxidant status in lipopolysaccharide and diclofenac-induced liver damage[154].Vitamin E is a fat-soluble vitamin and one of the most potent antioxidants.This action is attributed to the ability of the hydroxyl group to scavenge free radicals and restoration of GSH levels and hence the improvement of oxidant/antioxidant status.Accordingly,in addition to other mechanisms,vitamin E effectively reduces inflammation[155] but not fibrosis[156].
Nevertheless,a recentin vivostudy by Aljuhret al[157] showed that using vitamins E and C loaded on selenium nanoparticles effectively reduces the induced hepatocellular damage,making it a potent combination for preventing and treating HCC.Acute hepatotoxicity induced by APAP overdose is typically countered by N acetyl cysteineviaits antioxidant activity and increasing the level of GSH in the liver[158].In cases of APAP-induced acute liver injury,N acetylcysteine is the antidote for hepatotoxicity as it can preserve GSH stores and counteract the toxic metabolite NAPQI[159].In addition,N acetyl cysteine exerted favorable effects at increasing GSH peroxidase and decreasing oxidative stress in liver fibrosis induced by carbon tetra chloride[160].Mitoquinone (MitoQ),mitochondrial-targeted coenzyme Q,is a recent advance in antioxidant therapy that delivers coenzyme Q directly to the mitochondria[161].In carbon tetrachloride-induced liver fibrosis,MitoQ showed a reduction in lipid peroxidation marker,4-hydroxynonenal,in vivoand inhibition of cultured HSC activation[162].Accordingly,MitoQ seems promising in mitigating liver fibrosis,but further studies are needed to confirm its efficacy.
Anti-fibrotic and anti-inflammatory drugs
Both acute and chronic liver disorders involve a series of cytokine and chemokine production and inflammatory cell infiltration that promote fibrogenesis[163,164].This emphasizes the importance of using anti-fibrotic and anti-inflammatory drugs to modulate fibrogenesis and reduce the progression of liver fibrosis.Pirfenidone is a pyridone derivative with antifibrotic and anti-inflammatory properties and is mainly used for pulmonary fibrosis[165].These actions are attributed to the ability of pirfenidone to suppress TGF-β and NF-κB activation and thus decrease inflammatory cell infiltration and excess matrix deposition.Along with the antioxidant activity of pirfenidone,it effectively diminishes liver fibrosis[166],as shown in a two-yearin vivostudy on CHC virus-infected patients[167].Statins and anti-NADPH oxidases as anti-fibrotic classes,peroxisome proliferator-activated receptor alpha modulators,and timolumab as immunomodulators have been recently investigated and declared promising for decreasing inflammation and fibrosis in cases of primary sclerosing cholangitis[168].
Immunosuppressants
Autoimmune hepatitis is a chronic inflammatory liver disease that occurs when helper T cells and cytotoxic T cells attack the liver causing inflammation that may progress to liver cirrhosis.Autoimmune hepatitis can be acute,fulminant,or chronic and,like other autoimmune disorders,require immunosuppressive therapy to suppress the disease progression.The treatment of autoimmune hepatitis involves using corticosteroids as antifibrotic agents and azathioprine,and when this line of management is insufficient,mycophenolate mofetil and calcineurin inhibitors are used[169,170].In contrast,these drugs require more investigation for their use in other autoimmune liver disorders,such as primary sclerosing cholangitis and primary biliary cirrhosis[171].
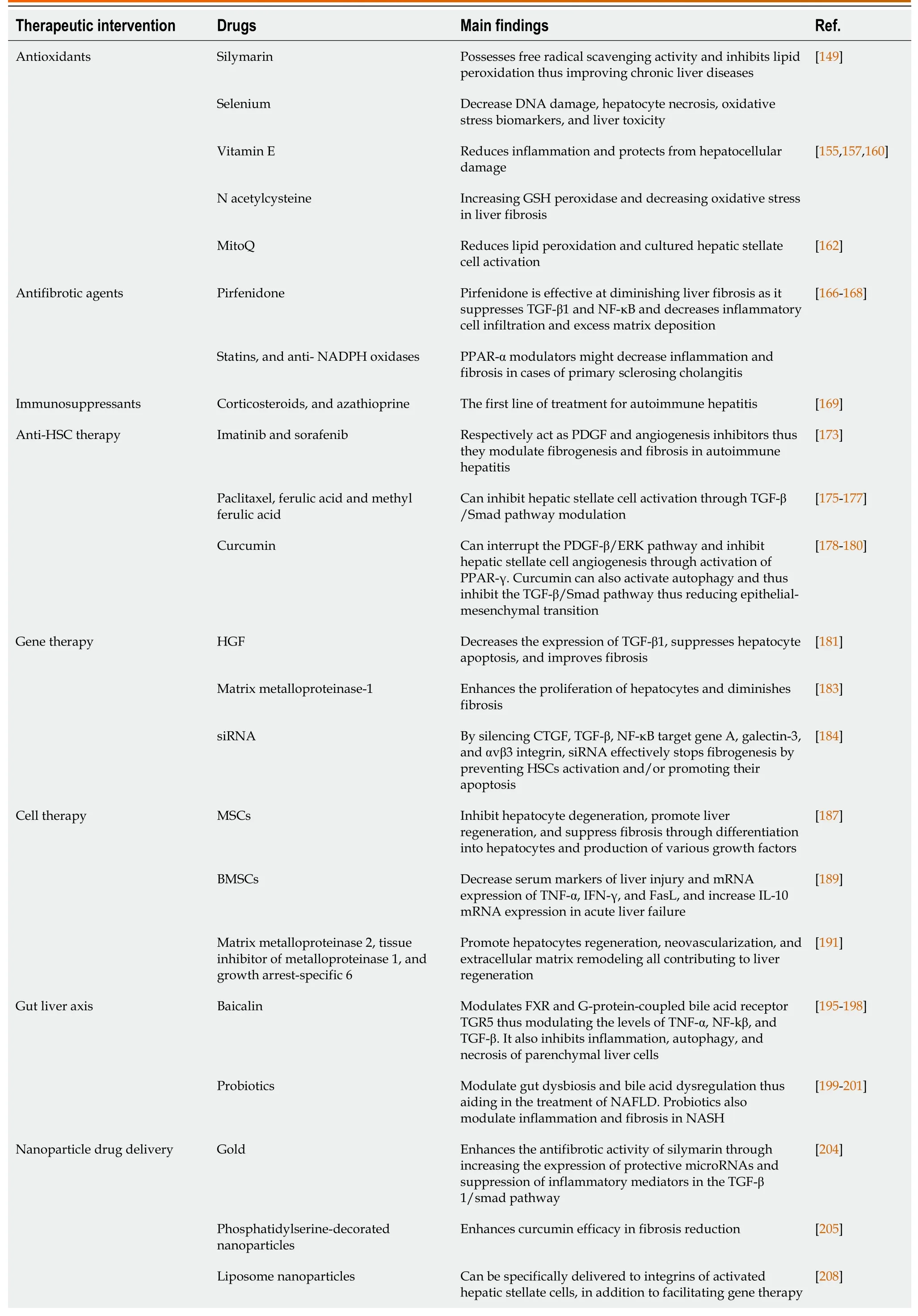
Table 2 Therapeutic interventions implicated in acute and chronic liver disorders
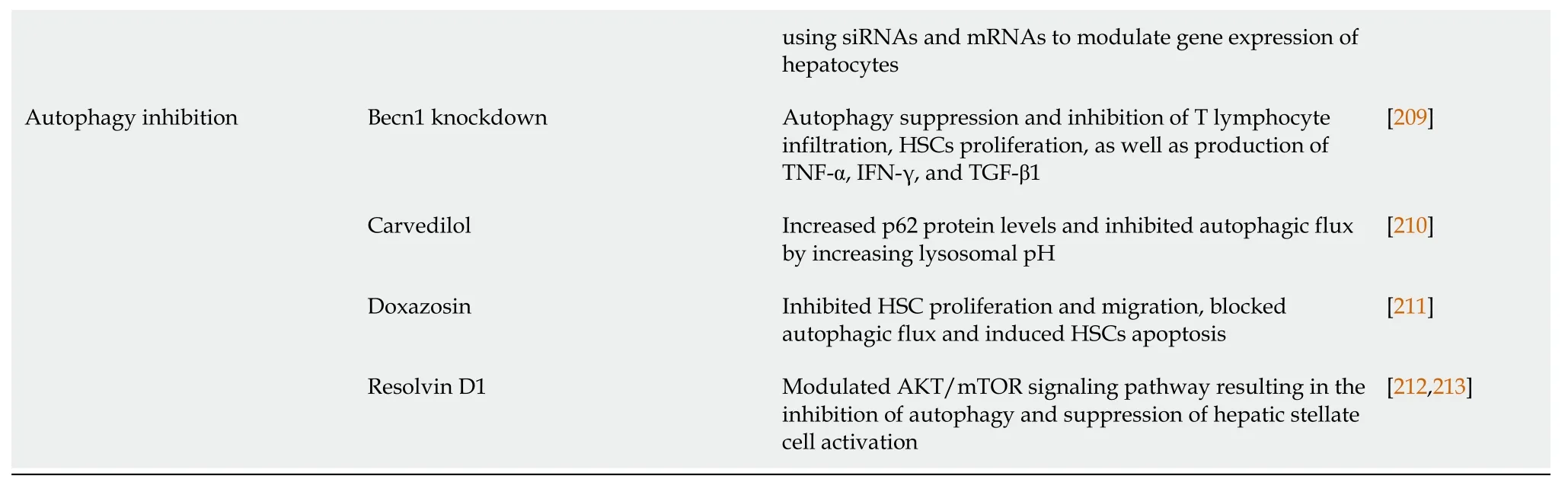
GSH: Glutathione;PPAR-α: Peroxisome proliferator-activated receptor alpha;CTGF: Connective tissue growth factor;TGR5: G-protein-coupled bile acid receptor;MitoQ: Mitoquinone;HSCs: Hepatic stellate cells;TGF-β: Tumor growth factor-beta;NF-κB: Nuclear factor-kappaB;PDGF: Platelet-derived growth factor;HGF: Hepatocyte growth factor;ERK: Extracellular signal-regulated kinase;siRNA: Small interfering RNA;mTOR: Mechanistic target of rapamycin;TNF: Tumor necrosis factor;IFN: Interferon;NAFLD: Non-alcoholic fatty liver;NASH: Non-alcoholic steatohepatitis;FXR: Farnesoid X receptor;IL: Interleukin;MSC: Mesenchymal stem cell;BMSC: Bone marrow-derived mesenchymal stromal cell.
Anti-HSCs therapy
One of the most important mitogens in profibrogenic HSC activation following liver damage is PDGF[172].A recent study using PDGF and angiogenesis inhibitors as imatinib and sorafenib,respectively,concluded that they were able to modulate fibrogenesis and fibrosis in induced autoimmune hepatitis models[173].Silymarin possesses antioxidant activity and antifibrotic properties through inhibition of KCs activation,decreasing extracellular matrix deposition,and inhibiting the production of IL-1 and IL-8 on HSCs[174].As TGF-β is a crucial cytokine for HSC fibrogenesis and hence liver fibrosis progression[172],different studies have been conducted to study the effect of various substances to obstruct TGF-β/Smad signals.In vitrostudies on paclitaxel,ferulic acid,and methyl ferulic acid were encouraging for inhibition of HSC activationviaTGF-β/Smad pathway modulation[175-177].Curcumin is a natural antioxidant,anti-inflammatory,and antifibrotic agent that can modulate different apoptotic pathways during tissue injury.Recent studies showed that curcumin could interrupt the PDGF-β/ERK signaling pathway and inhibit HSC angiogenesis by activating PPAR-γ[178,179].Furthermore,curcumin can activate autophagy and thus inhibit the TGF-β/Smad pathway,which reduces epithelial-mesenchymal transition[180].Accordingly,curcumin is considered a good candidate for treating liver fibrosis.
Gene therapy
Acute liver injury is usually reversible;however,chronic liver damage is a progressive condition that usually progresses from inflammation and fibrosis to cirrhosis.That is why extensive investigations on gene therapy have been conducted with various genes and delivering vectors to modulate liver fibrosis and cirrhosis.Hepatocyte growth factor (HGF) is an essential antiapoptotic and hepatoprotective factor for hepatocytes and an antifibrogenic agent in liver fibrosis models.HGF gene therapy has been studied for liver cirrhosis in rats and was shown to decrease the expression of TGF-β,suppress hepatocyte apoptosis,and improve fibrosis in dimethyl nitrosamine-induced cirrhosis[181].Due to the ability of HGF to suppress TGF-β,it exhibits immunomodulatory action that is promising in cases of autoimmune disorders,but further investigations are still required[182].As HSCs generate abundant amounts of extracellular matrix during fibrogenesis,matrix metalloproteinase-1 delivered by adenovirus to fibrotic livers enhances the proliferation of hepatocytes and diminishes fibrosis[183].Another mechanism involves the use of small interfering RNA (siRNA) to silence the genes that are essential for the process of fibrosis,such as connective tissue growth factor,TGF-β,NF-κB target gene A,galectin-3,and αvβ3 integrin.Silencing these genes stops fibrogenesis effectively by preventing HSCs activation and promoting their apoptosis[184].
Cell therapy
Stem cells are a category of cells that can replicate and differentiate into numerous types of specialized cells in the body[185].During the last two decades,stem cell-based therapy has been extensively investigated and appears promising for liver regeneration.Thus,it is a considerable alternative for liver transplantation and overcoming its demerits like the shortage of liver donors,high cost,and surgical complications.Various types of stem cells have been studied in acute and chronic liver disorders,including embryonic stem cells,induced pluripotent,and adult stem cells composed of the liver,mesenchymal,and hematopoietic stem cells[186].Mesenchymal stem cells (MSCs) are a suitable alternative for liver transplantation because they inhibit hepatocyte degeneration,promote liver regeneration,suppress fibrosisviadifferentiation into hepatocytes,and produce various growth factors[187].
Moreover,combining MSCs with induced bone marrow-derived macrophages showed stronger antifibrotic activity and hence better improvement of the cirrhotic liver than monotherapy[188].Anin vivostudy investigating cell therapy in mice used four types of cells;mature hepatocytes,fetal liver cells,bone marrow-derived mesenchymal stromal cells (BMSCs),and induced hepatic stem cells for concanavalin A-induced fulminant hepatitis causing ALF and fumarylacetoacetate hydrolase-deficient induced chronic liver failure.Remission of concanavalin A-induced ALF was only noticed with BMSCs as they decreased serum markers of liver injury and mRNA expression of some inflammatory cytokines,including TNF-α,IFN-γ,and FasL,and increased IL-10 mRNA expression.In the chronic liver failure model,mature hepatocytes in the adult liver were the most effective for liver regeneration compared to other cell types.However,these hepatocytes are not common in clinical applications due to their limited sources[189].In acute liver injury induced by carbon tetra chloride,using hepatocyte-like cells derived from embryonic stem cells showed the potential for attenuation of liver injury and the remission of induced liver fibrosis[190].Furthermore,rather than cell transplantation,using trophic factors such as matrix metalloproteinase 2,tissue inhibitor of metalloproteinase 1,and growth arrest-specific 6,released from embryonic-derived hepatocyte-like cells,promoted hepatocytes regeneration,neovascularization,and extracellular matrix remodeling,all of which contribute to liver regeneration[191].Despite the numerous advantages of stem cell therapy,safety concerns such as ethical approval of embryonic stem cell use,lack of knowledge of appropriate transmission methods,enhancement of tumor growth,and incomplete prediction of tissue response are limiting their use nowadays[192].
Gut-liver axis
The relationship between the gut and the liver involves the delivery of intestinal contents to the liver through the portal vein and the transport of bile acids and immunoglobulins from the liver back to the intestines.Any disruption of the homeostasis of this axis through altering gut microbiota (gut dysbiosis),bile acid composition,or intestinal barrier damage will result in the exposure of the liver to these microbes and their metabolites which is critical in the pathogenesis of the ALD,NAFLD and even liver cirrhosis[193,194].That is why various experiments and clinical trials targeting the gut-liver axis are being studied to treat liver disorders,including NASH,NAFLD,and chronic hepatitis B and C[195].The farnesoid X receptor (FXR) is a nuclear receptor highly expressed in the gut-liver axis and regulates bile acid production,detoxification,maintenance of triglyceride homeostasis,and enhancement of the function of the intestinal epithelial barrier[194,196,197].Baicalin is a natural flavonoid studied on various liver disorders and exhibited favorable effects such as inhibition of inflammation and autophagy and necrosis of parenchymal liver cells,thus decreasing liver injury.One of the pathways involved in baicalin effects is FXR and G-protein-coupled bile acid receptor,as they can modulate TNFα,NF-kβ,and TGF-β levels[195,198].As gut dysbiosis and bile acid dysregulation are directly related to NAFLD’s pathogenesis,using various probiotics,prebiotics,and synbiotics has been proven to be promising for treating NAFLD[199,200].In addition to the role of probiotics in NAFLD and their ability to modulate inflammation and fibrosis in NASH,probiotics are an attractive target for gut-liver-related disorders as they are also cost-efficient,with mild adverse effects and nearly no long-term adverse reactions[201].
Nanoparticle drug delivery
Recently,nanomedicine gained much attention as an innovative way for effective drug delivery in various resistant types of diseases.Numerous nanoparticle types are used in liver fibrosis treatment:Inorganic oxides and metals[202] or organic micelles and liposomes[203].Gold,an inert inorganic widely used material,is formulated in nanoparticle form to deliver silymarin to fibrotic livers induced by carbon tetrachloride.This process enhanced the antifibrotic activity of silymarin,attributed to increased expression of protective miRNAs and suppression of inflammatory mediators in the TGF-β/Smad pathway[204].As we previously mentioned,the anti-fibrotic action of curcumin,enhancing drug delivery and bioavailability of curcumin using phosphatidylserine-decorated nanoparticles,further enhances curcumin efficacy in fibrosis reduction[205].
Interestingly,nanoparticles can also target different liver cells involved in liver fibrosis.As the expression of c-x-c chemokine receptor 4 (CXCR4) and VEGF is associated with HSCs activation and hence liver fibrosis progression,combining CXCR4 antagonist in nanoparticles with siRNA against VEGF provided significant inhibition of the process of angiogenesis making it auspicious treatment for liver fibrosis[206].Considering liposome nanoparticles,the use of liposomes to be specifically delivered to integrins of activated HSCs rather than any other type of liver cells has been conducted,making it available to deliver therapeutic drugs to special sites overcoming their complications[207].A novel advantage of liposome nanoparticles is that they facilitate gene therapy using siRNAs and mRNAs to modulate gene expression of hepatocytes instead of using viruses as carriers[208].
Autophagy
As we have mentioned,the BECN1 protein has been involved in autophagy,resulting in ferroptotic cell death.A study on knocking down BECN1 showed inhibition of autophagy and its consequent inflammation in addition to increasing prostaglandin E2 (PGE2) levels.Modulation of the prostaglandinendoperoxide synthase 2/PGE2 pathway may cause suppression of HSC proliferation and lymphocyte infiltration,all contributing to MSCs’ enhanced antifibrotic activity[209].That is why inhibition of autophagy is a potential target for liver fibrosis treatment.Carvedilol,a non-selective B-blocker,has been thought to possess antifibrotic activity.Testing this theoryin vitrorevealed that carvedilol can alleviate liver fibrosis by inhibiting the autophagy of HSCs and enhancing their apoptosis[210].Doxazosin,an alpha-1 adrenergic receptor agonist,has also been studiedin vitroandin vivoand showed similar action to carvedilol on activating apoptosis of HSCs and inhibiting autophagy through the PI3K/Akt/mTOR signaling pathway[211].Resolvin D1 is a polyunsaturated fatty acid that has been proven effective in various liver disorders,such as acute liver injury and liver fibrosis,due to its antioxidant,anti-inflammatory,and antifibrotic effects.Further investigations on resolvin D1 on CCL4-induced liver fibrosis demonstrated its ability to modulate the AKT/mTOR signaling pathway,resulting in inhibition of autophagy and suppression of HSC activation,which further intensifies resolvin D1 liver protective effect[212,213].
CONCLUSION
Collectively,acute and chronic liver diseases are worldwide problems with multifactorial pathogenesis.The exact pathological mechanism of several liver disorders is still unclear.However,many suggested mechanisms are involved,including but not limited to oxidative stress,inflammation,autophagy,and miRNA.The role of autophagy and miRNA is still unclear and requires more clarification.Besides,it may be a new way to find new therapy for hepatic disorders.Recent therapeutic strategies like gene therapy,stem cell therapy,gut microbiota,and even nanoparticle formulations require more investigations and improvements.
FOOTNOTES
Author contributions: Ali FE and Bakr AG designed and critically wrote the manuscript;Abd El-Aziz MK and Sharab EI collected data and drafted the manuscript.
Conflict-of-interest statement: All the authors report no relevant conflicts of interest for this article.
Open-Access: This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is noncommercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin: Egypt
ORCID number: Fares EM Ali 0000-0001-8341-7458;Mostafa K Abd El-Aziz 0000-0003-0252-5065;Elham I Sharab 0000-0001-9147-8373.
S-Editor: Wang JJ
L-Editor: A
P-Editor: Wang JJ
杂志排行

World Journal of Hepatology的其它文章
- Current therapeutic modalities and chemopreventive role of natural products in liver cancer: Progress and promise
- Acute-on-chronic liver failure in patients with severe acute respiratory syndrome coronavirus 2 infection
- Detection of colorectal adenomas using artificial intelligence models in patients with chronic hepatitis C
- Prognostic role of ring finger and WD repeat domain 3 and immune cell infiltration in hepatocellular carcinoma
- Rising incidence,progression and changing patterns of liver disease in Wales 1999-2019
- Influence of non-alcoholic fatty liver disease on non-variceal upper gastrointestinal bleeding: A nationwide analysis