饮用水中超痕量多环芳烃测定方法的优化
2023-03-14王怿婷
王怿婷
(北京市自来水集团有限责任公司水质监测中心,北京 100192)
多环芳烃(PAHs)是一类对人体具有较强的毒性、突变性和致癌性的有机污染物,主要导致胃癌、肺癌和皮肤癌[1]。由于工业上石油、煤等原料的大量使用,PAHs在环境中已广泛存在[2],也是水体中重点监测的污染物之一。PAHs种类繁多,美国环保署(EPA)将其中的16种PAHs列为优先检测物[3],中国将其中的7种PAHs列为优先控制污染物[4]。我国的饮用水卫生标准中规定水中苯并[a]芘含量应小于0.01μg/L,多环芳烃总量不得大于2μg/L[5]。水体中的多环芳烃含量为痕量级[6- 7],因此对于水体中PAHs的富集浓缩和更有效的提高检测灵敏度显得尤为重要。目前水中多环芳烃类物质的前处理主要采用液液萃取、固相萃取法,其分析方法主要有气相色谱法[8]、气相色谱质谱法[9]、高效液相色谱法等[10- 12],应用最多的是固相萃取-液相色谱法(HPLC),该方法稳定、适用性广、操作便捷,但对PAHs多种混合物的分析过程较长,不能满足对该污染物的快速筛查和分析。本研究针对荧蒽、苯并(b)荧蒽、苯并(k)荧蒽、苯并(a)芘、苯并(g,h,i)苝、茚并(1,2,3-cd)芘6种被列为优先控制的PAHs,通过对比常规HPLC测定方法,并优化色谱参数及分离条件,建立了一种更为快速、高效的PAHs超高效液相色谱(UPLC)分析法,更有效地提高了检测灵敏度和精密度,满足实际水样中PAHs的痕量监测要求。
1 实验部分
1.1 仪器和试剂
仪器:Waters 2695型液相色谱仪(HPLC),配2475荧光检测器,色谱柱为PAHs专用柱(4.6mm×150mm,5μm);Waters UPLC-H Class超高效液相色谱仪(UPLC),配FLR荧光检测器,色谱柱为PAHs快速柱(4.6mm×50mm,3μm)。
试剂及材料:甲醇、乙腈(美国Fisher公司,色谱纯)、二氯甲烷,Milli-Q超纯水;Waters HLB固相萃取小柱(6mL,200mg);6种多环芳烃物质:荧蒽、苯并(b)荧蒽、苯并(k)荧蒽、苯并(a)芘、苯并(g,h,i)苝、茚并(1,2,3-cd)芘混合标准溶液(200μg/mL,美国AccuStandard公司)。
1.2 实验方法
1.2.1仪器参数及色谱条件
常规测定(HPLC)色谱条件:Waters 2695-HPLC,检测器为2475荧光,PAHs色谱柱(4.6mm×150mm,5μm),柱温30℃,流动相为纯甲醇,等度洗脱,流速1mL/min,进样量10μL,荧光检测器波长参数见表1。
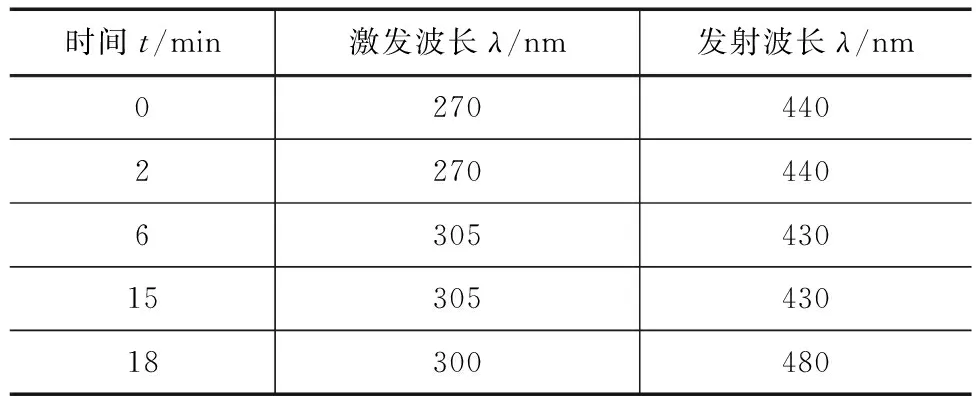
表1 HPLC荧光检测器波长参数
快速测定(UPLC)色谱条件:Waters H-Class-UPLC,检测器为FLR荧光,PAHs快速柱(4.6mm×50mm,3μm),柱温30℃,进样量5μL,流动相为乙腈、水,梯度洗脱,洗脱条件见表2,荧光检测器波长见表3。
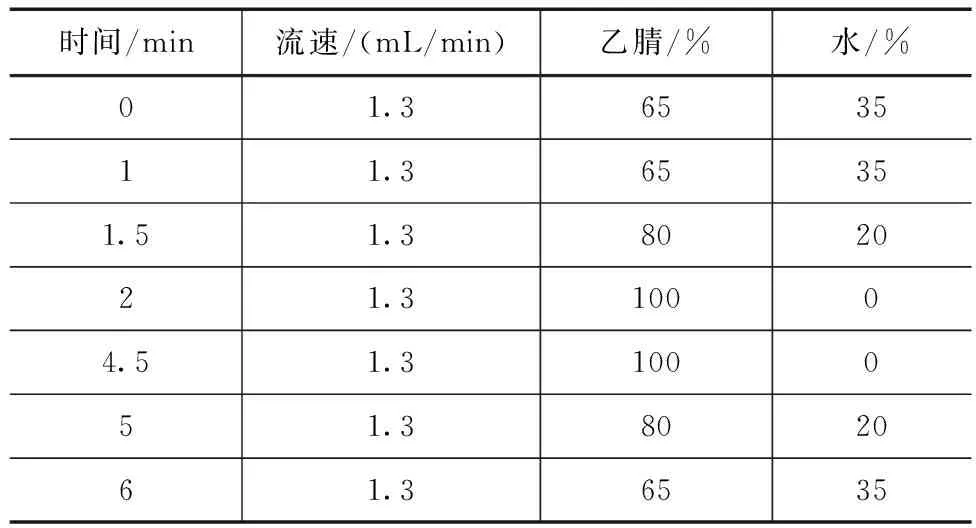
表2 UPLC梯度洗脱条件
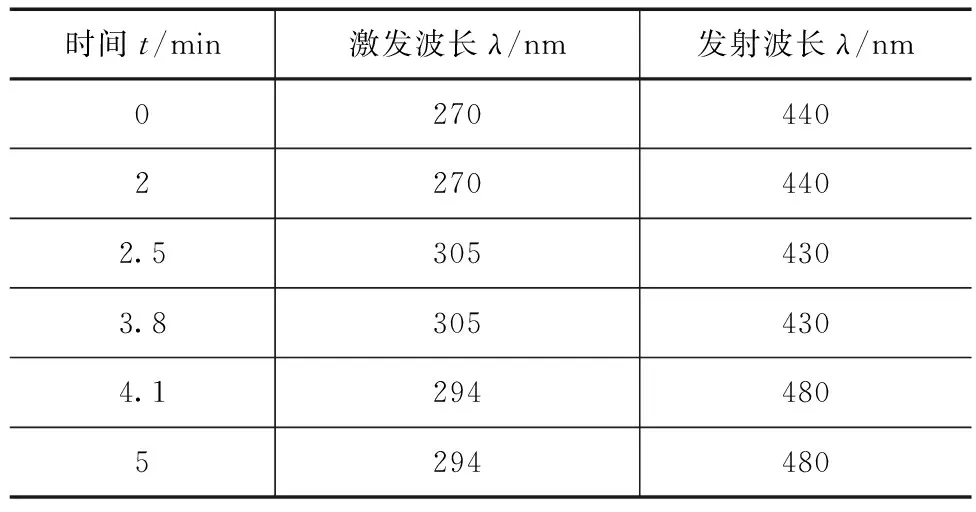
表3 UPLC荧光检测器波长参数
1.2.2样品前处理
对于水体中痕量级的多环芳烃,本实验采用固相萃取法富集,将Waters HLB固相萃取小柱依次用二氯甲烷5mL、甲醇5mL、纯水10mL进行充分活化,在1L水样中加入10mL甲醇和5g氯化钠混匀,并以10mL/min的流速进行上样富集,完成后用5%甲醇水溶液清洗杂质,用2mL二氯甲烷洗脱待测物质,3次洗脱后收集洗脱液并吹干,用乙腈定容至1mL待测。
2 结果与讨论
2.1 分析条件探讨
2.1.1色谱柱分析与探讨
分别选取PAHs专用柱(4.6mm×150mm,5μm)和PAHs快速柱(4.6mm×50mm,3μm)进行实验比对,分别按照1.2.1中的常规测定(HPLC)和快速测定(UPLC)的色谱条件洗脱分离,6种多环芳烃分离图谱如图1—2所示,2种测定方法的保留时间对比见表4。结果表明通过PAHs快速柱及UPLC建立的6种PAHs分析法可在7min内完成对混合标准物质的有效分离与测定,大大提高了检测效率,检测进程的缩短也更有利于节约溶剂与标准品、进样量减少、环保性更佳。这主要是由于选择更小粒径的柱子,其填料更加紧密均一,柱效更高、速度更快。需要注意的是PAHs快速柱由于长度只有50mm,洗脱时应采用梯度洗脱,等度洗脱分离效果不佳,洗脱条件见表2。
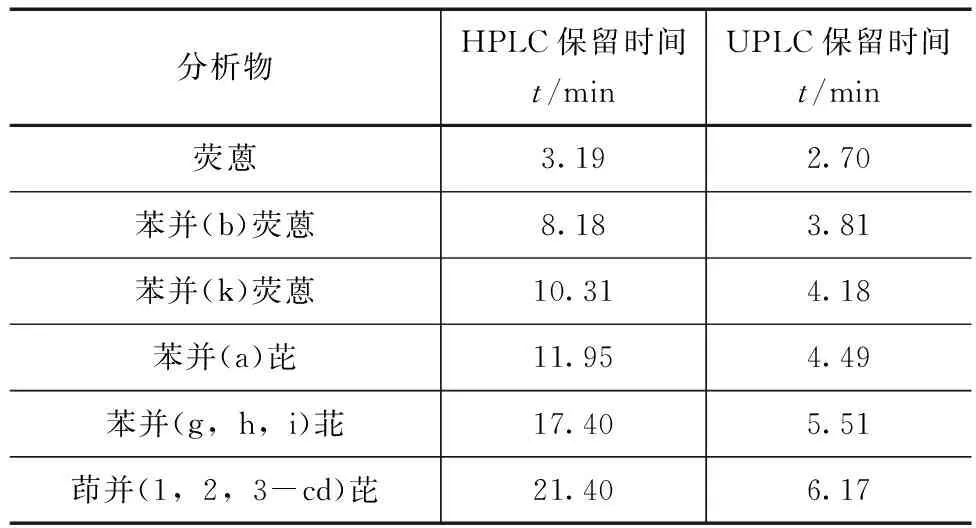
表4 HPLC与UPLC测定6种PAHs的保留时间比较
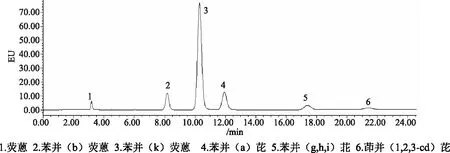
图1 6种多环芳烃HPLC荧光色谱图

图2 6种多环芳烃UPLC荧光色谱图
2.1.2荧光波长参数的优化
检测的6种多环芳烃在紫外区也有吸收,但检测灵敏度较低,故本实验选用响应值更高的荧光检测器分析测定。为了使6种PAHs化合物达到更好的灵敏度和分离度,经过反复实验优化,比较了6种多环芳烃物质在不同波长下的出峰时间和灵敏度,最终确立了最佳波长参数,见表1—3。需要说明的是波长参数变动较大时可造成出峰时间和基线漂移、波动较大,因此对于检测波长接近的物质可选用一致的激发与发射波长。另外,相对于其他5种PAHS来说,茚并(1,2,3-cd)芘对于荧光波长参数的变动最为敏感,通过多次实验得出茚并(1,2,3-cd)芘的最佳激发、发射波长分别为294、480nm。
2.2 线性范围与检出限
选用优化后的快速测定PAHs的UPLC法,进行方法的有效性验证。取6种PAHs混合标准物质用甲醇稀释,配制成浓度为1.0、2.5、5.0、10、25、50μg/L的混合标准系列,以浓度为横坐标、峰面积为纵坐标建立标准曲线,其线性范围和回归方程见表5。结果表明,6种PAHs在1~50μg/L范围内线性良好,相关系数均大于0.999,满足日常测定要求。该方法检出限通过公式MDL=t(n-1,0.99)×S得出(式中MDL:方法检出限;n:添加低浓度样品的平行测定次数;t:自由度为n-1、置信度为99%时的t分布;S∶n次平行测定的标准偏差)。故选用浓度为1.0μg/L的标准溶液,平行测定七次,根据上述公式计算出方法检出限,结果见表5,6种多环芳烃化合物的方法检出限(MDL)在0.30~0.70ng/L范围内,适用于水体中PAHs的痕量分析。
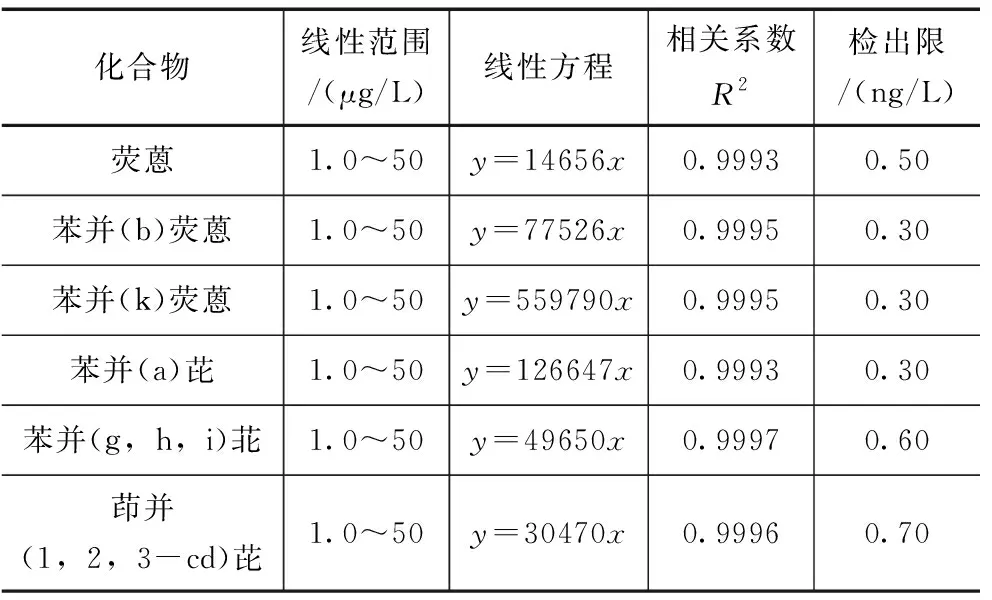
表5 6种PAHs的线性范围及检出限
2.3 精密度与准确度
按照提速增效后的UPLC配置PAHs快速柱的实验方法分别对不含此6种PAHs的空白水样进行高低浓度加标回收实验,加标浓度为1.0μg/L和50μg/L,平行测定6次,各物质的精密度和回收率见表6。结果显示高低浓度加标后的回收率为83.3%~94.2%,相对标准偏差(RSD)小于7.2%,该方法的准确度、精密度均能满足水样中6种PAHs的快速痕量分析要求。
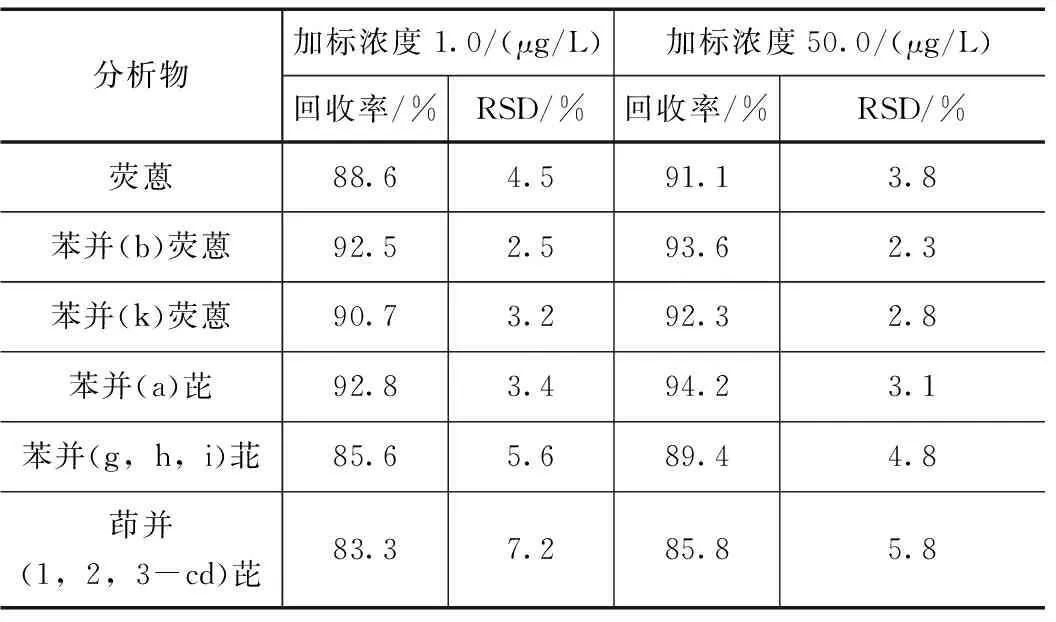
表6 6种多环芳烃的加标回收率与精密度
2.4 实际水样测定
该方法经过有效性验证后,选取某市有代表性的几个水厂出水、原水及管网水进行分季度的PAHs监测普查工作,测定结果显示在各批次水样中6种PAHs均未检出。
3 结语
本研究以6种被列为优先控制的多环芳烃为例,通过选取PAHs快速专用色谱柱,优化实验条件,建立了一套固相萃取-超高效液相色谱法(UPLC)检测水中的PAHs。该方法极大地缩短了分析过程,可在7min内完成6种PAHs的分离与测定,提升了大规模样本的检测通量,方法稳定,线性良好,重现性及灵敏度高,同时由于进样量的减少,更利于节约标准参照物成本、节省溶剂、减少毒性。提速增效后的UPLC方法也是未来色谱分析的发展趋势,其应用前景良好。该法适用于饮用水中PAHs的痕量测定要求,但目前检测种类有限,对于除6种之外的其余多种PAHs的快速测定也是后续优化研究的方向。
