金电极与吡啶末端连接界面结构的力学变化过程理论研究*
2023-03-05刘琳孙峰李雨晨严岩刘冰心羊志邱帅李宗良
刘琳 孙峰 李雨晨 严岩 刘冰心 羊志 邱帅 李宗良
(山东师范大学物理与电子科学学院,济南 250358)
利用基于第一性原理的绝热拉伸模拟方法计算了4,4′-二吡啶分子与不同构型金电极之间的作用过程,研究了分子在外力作用下逐渐远离金电极过程中分子与电极间界面结构特有的演化过程以及体系能量与作用力的变化特征.结果显示,分子在远离锥形电极过程中很容易出现近于垂直地连接到锥形电极第二层金原子上的特有连接构型,同时由于吡啶末端的排斥作用,电极尖端的金原子偏向一侧.分子从第二层金原子上断开并连接到尖端金原子上需要1.3—1.5 nN 的拉力作用,明显大于分子从尖端电极上断开所需要的0.8—1.0 nN的作用力,从而揭示了实验中二吡啶分子结在形成过程中作用力与界面构型变化之间的对应关系.4,4′-二吡啶分子与平面金电极的作用较弱,只需要不到0.5 nN 的作用力就可断开,而当分子连接到吸附在平面电极表面的孤立金原子上时,可以承受约1.7 nN 的作用力.以上研究表明基于第一性原理的绝热拉伸模拟方法不仅可以揭示分子与电极之间的界面结构演化过程,而且通过作用力的计算可以很好地识别实验中分子与电极间的特有界面结构.
1 引言
在纳米或分子尺度上构筑功能电子器件一直是人们不断追求的目标[1-6].随着纳米技术的不断提高,人们已能够对单个分子进行操控和测量,并且发现单个分子或者纳米结构具有非常好的光电子学功能特性,如开关[6-9]、整流[10-13]、存储[14-16]、传感器[16-20]、逻辑门[5]或者场效应管[21-24]等功能.目前,精准操控单个原子或分子还具有极大的挑战,这一目标不仅关系到能否实现分子尺度器件的应用,也是突破和超越纳米和分子尺度“卡脖子”技术的关键[25].单分子结组装技术为实现分子尺度的识别、控制及研究原子分子间的相互作用建立了可行的研究平台[2,26-28],但完全通过实验仍然无法直接完成这一任务,必须发展相关理论方法并辅助相关理论计算和分析才能有效地实现分子尺度的识别与控制[29-31].
分子结是由两纳米电极与连接在两电极之间的功能分子构成的[32,33].为了让分子与电极间形成较稳定的连接,一般采用具有特定末端的分子构建分子结.常用的分子末端有硫醇/酚末端[34-+]、氨基末端[40-+]和吡啶基末端[46-+],其中,吡啶基末端是一种非常值得关注的分子末端.实验发现以吡啶基为末端的分子结在形成过程中经常出现显著的高低电导转换现象[29,30,49],利用这一现象可以将分子设计成分子开关或者分子传感器.然而从以前的研究中可以发现,对于这一现象产生的物理机制和具体物理过程还存在很多困惑.许多研究认为随着电极距离变化,分子与电极表面间出现不同夹角,从而导致分子结出现高低电导转换现象[29,30].虽然按照文献中给出的电极界面构型,能够说明分子相对于电极的夹角变化可以使体系出现高低电导转换现象,但却无法解释分子结由高电导向低电导转换过程中所需的拉力明显大于分子结从低电导状态断裂时所需的作用力[29,50],更甚至,有的文献在电极作用力上给出了与实验完全相反的计算结果[30].另外,电极作用力和电导在此过程中产生的跳跃式变化用分子与电极间的夹角变化也无法理解.研究发现,以吡啶基为末端的分子结在拉伸过程中出现的高电导,是由于吡啶基中的N 原子在分子结拉伸过程中可以近乎垂直地连接到电极尖端的第二层金原子导致的[51,52].当N 原子连接到第二层金原子上时,可以承受约1.2 nN 的作用力,略小于实验测量结果,此时电极尖端的金原子在吡啶基的作用下偏向一侧.而进一步拉伸分子结,N 原子从第二层金原子跳转到尖端金原子上,分子结的电导出现接近一个数量级的显著降低,从而产生高低电导转换现象.分子从电极尖端断裂所需的作用力约为0.8 nN,与实验测量结果完全一致.由于采用了基于第一性原理的分子结拉伸计算,计算量较大,在以前的研究中我们只采用了理想的三棱锥形电极模拟实验中的探针电极.为了从分子与电极作用力的角度验证这一结果的普遍性与可靠性,下面进一步研究4,4′-二吡啶分子与不同构型电极之间的作用过程,利用基于第一性原理的拉伸模拟方法计算分子与不同构型电极间能够承受的作用力,并揭示分子与电极之间作用过程中电极界面结构的演化过程,从而通过作用力的计算实现分子与电极间界面的识别[52-+].
2 理论模型与计算过程
扫描隧道显微裂结(STM-BJ)技术和力学可控裂结(MCBJ)技术是组装分子结的常用实验技术.在分子结组装过程中,分子可以与电极形成不同的连接方式.为了重点讨论分子与电极间作用力及界面结构的变化关系,在下面的研究中采用了单电极模型.计算显示,采用单电极模型对分子与电极间的作用力及界面结构的影响很小,可以很好地计算分子与电极间的作用力及关键界面结构,特别是对于部分需要考虑更多原子的电极构型,可以有效地提高计算效率.与双电极模型相比,采用单电极模型计算的最大拉力的偏差一般小于0.1 nN,另外分子上端位置在单电极模型中因完全固定,与双电极模型也存在一些小的差异.
分子与电极连接的初始结构如图1 所示,其中图1(a)是分子初始吸附在理想的锥形电极旁边(体系I);图1(b)中的电极只有最尖端两层金原子构成锥形,而第三层金原子明显多于体系I 中的第三层金原子并形成平台状结构(体系II);图1(c)中的电极模拟了平面电极表面上吸附有孤立金原子的情形(体系III);图1(d)中的电极初始为平整的平面电极(体系IV).
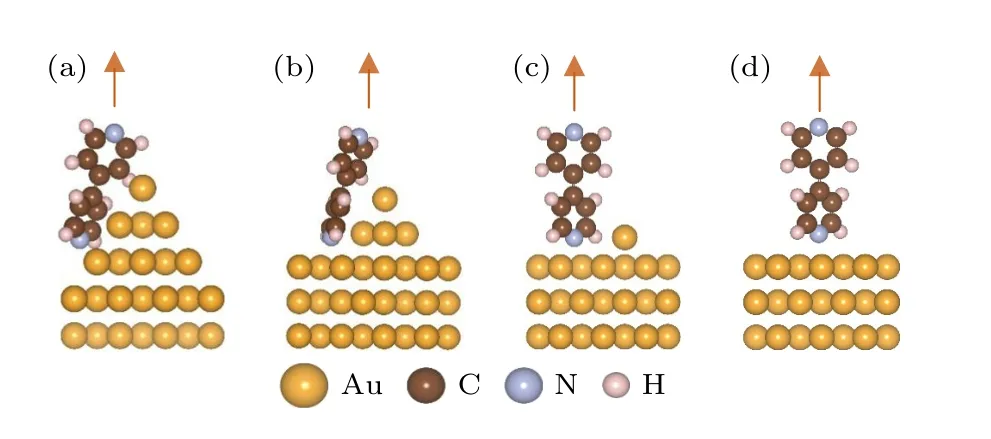
图1 4,4′-二吡啶分子连接到不同构型金电极上的初始结构 (a) 分子连接在理想锥形电极旁边(体系I);(b) 分子与只有最尖端两层金原子构成锥形的电极相连接,电极的第三层有较多金原子形成平台构型(体系II);(c) 分子与吸附有孤立金原子的平面电极相连接(体系III);(d) 分子与平面电极相连接(体系Ⅳ)Fig.1.The initial configurations for 4,4′-bipyridine connecting with different gold electrode: (a) The molecule is connected beside ideal pyramid-shaped gold electrode (system Ⅰ);(b) the molecule connects with the gold electrode in which only the top two layers of Au atoms form pyramid shape (system Ⅱ).In this system the third layer of the electrode contains more Au atoms which form a platform;(c) the molecule adsorbs on planar-shaped gold electrode with a single Au atom on the electrode surface (system Ⅲ);(d) the molecule adsorbs on planar-shaped gold electrode without single surface Au atom (system Ⅳ).
为了模拟分子在外力牵引下与电极相互作用的过程,计算中固定分子最上端的N 原子以模拟外力(即另一电极)对N 原子的控制,而分子中其他原子的自由度均放开进行几何结构优化.对于电极,考虑到原子间相互作用的局域性,对跟分子近邻和次近邻的金原子进行几何结构优化,而最底层金原子及离分子较远的部分金原子在计算中固定不动,这样处理不仅合理考虑了只有连接界面附近的金原子在分子作用下会出现明显移动而大多数金原子受块体金电极晶格限制不能自由移动的特点,同时可以有效地降低计算量以及计算有限体系产生的不必要的边界效应.分子在拉力作用下逐渐远离电极的过程是在上一步优化好的几何结构基础上,将上面的吡啶环和固定的金原子分别向上和向下移动一小段距离,然后再次对放开的原子进行几何结构优化.采取这一方式计算可以保证每一步结构优化都能对上一步结构存在记忆,因此能够很好地模拟分子在外力牵引下逐渐远离电极的绝热演化过程.体系几何结构优化选用LanL2DZ 为基矢,采用B3LYP 杂化交换关联泛函,在Gaussian16程序包上进行[55].
3 计算结果与讨论
由于金电极在断裂前一般都会出现整数倍G0(1 .0G0=2e2/h为单位量子电导)的标志性电导[2,26,29],因此不管是利用STM-BJ 技术还是用MCBJ 技术组装分子结,初始均借助于1G0电导的出现控制两电极从连接状态缓慢断开,以便更好地控制电极距离.在电极断开的过程中,由于两电极间的相互作用,MCBJ 装置中的两电极以及STM-BJ 装置中的探针电极容易形成如体系I 和体系II 的比较尖的锥形电极.电极断开后分子自组装到两电极之间.初始电极距离较小,分子将吸附在电极一侧(如图2(a)和图2(e)).随后分子在拉力作用下向上移动.图中显示,对于体系I,当分子上端的N 原子到电极的距离D=1.34 nm 时(图2 中D定义为上端的N 原子到最下面固定的金原子层的距离),4,4′-二吡啶分子将由吸附到锥形电极的侧面变为近垂直的吸附到尖端第二层金原子上,同时电极最尖端的金原子在分子的作用下偏向一侧(如图2(b)).随着分子继续向上移动,与下端N 原子相连的金原子也产生一定程度的上移.当分子上移到1.54 nm时,分子下端从第二层金原子上断开并连接到尖端金原子上,这时第二层中与N 原子相连的金原子缩回到原来的正常晶格位置,同时尖端的金原子也移回到中间位置(如图2(c)).当分子向上移动到1.68 nm 时,分子从尖端金原子上断开(如图2(d)).对于体系II,虽然第三层金原子形成平台结构,但由于尖端的两层金原子也形成了锥形结构,分子在上移过程中电极界面的变化与体系I 非常相似.与体系I 的主要不同表现在分子连接在电极第二层金原子时界面构型变化发生的距离略小于体系I(如图2(f)和图2(g)),这是由于第三层多出的金原子加强了对第二层金原子的束缚导致的.而当分子从尖端金原子断开时距离为1.72 nm (如图2(h)),略大于体系I 的断开距离,这表明由于第三层金原子的增多,第二层金原子与第三层金原子之间相互作用在增强的同时降低对尖端金原子的束缚作用,从而使尖端金原子有更大的弹性移动范围.对于体系III,分子初始连接在金电极的平面部分,距离表面孤立金原子的横向距离约为0.45 nm (如图2(i)),随着分子的上移,分子与电极平面部分的距离不断增加,与分子直接相连的金原子在分子牵引下也在一定程度上凸出金表面,但电极表面上的孤立金原子在此过程中移动很小 (如图2(j)).当分子移动到1.45 nm 时,电极表面孤立金原子迅速向左移动到相邻晶格空位上方,同时分子下端向右移动,并吸附到表面孤立金原子上 (如图2(k)).这一过程表明,当金电极表面存在孤立金原子时,吸附在孤立金原子附近的4,4′-二吡啶分子在一定距离下对孤立金原子具有很强的吸引作用,从而使孤立金原子在电极表面上移动,并最终吸附到表面孤立金原子上.将分子进一步拉到1.63 nm 时,分子从表面孤立金原子上断开(如图2(l)).对于体系IV,当分子与电极距离很小时,分子倾斜在电极表面上,随着距离的增大,分子下端相对电极表面产生滑移,并使分子逐渐变为直立.当D=1.41 nm 时,分子从平面电极断开.

图2 4,4′-二吡啶分子在拉力作用下远离不同构型电极表面的结构演化过程 (a)—(d) 体系I;(e)—(h) 体系II;(i)—(l) 体系III;(m)—(p) 体系IVFig.2.The structure evolution processes for 4,4′-bipyridine moves away from gold electrodes with different tip geometries: (a)—(d) System I;(e)—(h) system II;(i)—(l) system III;(m)—(p) system IV.
4,4′-二吡啶分子在拉力作用下远离不同构型电极过程中体系能量和作用力变化曲线如图3 所示.图3 中显示,对于体系I 和体系II,当分子连接到第二层金原子上后,能量曲线都出现了较深的势阱,其中体系I 中的势阱深约0.33 eV,而体系II 的阱深可达0.45 eV.这表明,拉动分子在电极侧面移动过程中,在适当距离下,分子很容易自发的近垂直的连接到第二层金原子上,特别是当第三层金原子较多时,分子跟第二层金原子作用更强.当分子从第二层金原子上断开并跳转到尖端金原子上时,体系I 的能量出现约0.55 eV 的较大幅度跳跃式下降,而体系II 的能量只出现了很小的跳跃式下降.这一差别是由于分子从第二层金原子上转移到尖端金原子上后,原来与分子相连的第二层上的金原子回缩的距离不同并释放不同的能量导致的.体系I 中的金原子在此过程中回缩了约0.24 nm,释放了较多的能量,而体系II 中的金原子只回缩了0.17 nm,因此只释放了较少的能量.对于体系I,分子连接到第二层金原子上的能量最小值比连接到尖端金原子上高约0.25 eV.但是对于体系II,能量只高了不到0.1 eV,这进一步表明第三层金原子的增多提高了对第二层金原子的束缚作用.第三层金原子对第二层金原子的束缚不同也导致了分子从能量最低点到与第二层金原子断开所移动距离不同,从图3(a)和图3(b)可以发现,对于体系I,分子移动距离约为0.13 nm,而对于体系II,这一距离约为0.11 nm.从分子与电极之间的作用力上看,分子从第二层金原子断开所需的拉力明显大于分子从尖端金原子上断开所需的拉力.对于体系I,分子从第二层金原子上断开所需的最大拉力约为1.3 nN,而从尖端金原子上断开所需的最大拉力为0.88 nN.对于体系II,分子从第二层金原子上断开所需的最大拉力约为1.4 nN,而从尖端金原子上断开所需的最大拉力为0.92 nN.这与实验上所测得的分子结断裂前先出现接近1.5 nN 的作用力,而后再以0.8—1.0 nN 的作用力断开完全一致[30,50].另外,分子从第二层金原子上断开到连接到尖端金原子上只经过了极短的距离变化,是一个突变式非弹性构型变化过程,这与实验中的作用力及电导发生的跳跃式突变是一致的.由于实验中作用力与体系电导具有同步变化的特点,因此根据我们的计算还可以推测,分子在远离锥型电极过程中出现的近于垂直地连接到第二层金原子上的特殊的界面构型,不仅是二吡啶分子结在拉伸过程中作用力发生由高到低的跳跃式变化的关键,同时也是体系电导产生由高到低跳跃式转换的原因.

图3 4,4′-二吡啶分子在拉力作用下远离不同构型电极过程中体系能量和作用力变化曲线 (a) 体系I;(b)体系II;(c) 体系III;(d) 体系IVFig.3.The energy and force traces for (a) system I,(b) system II,(c) system III and (d) system IV with 4,4′-bipyridine moving away from different gold electrodes.
对于体系III 和体系IV,分子在远离平面电极的过程中能量曲线同样出现了势阱,但势阱的深度明显小于体系I 和体系II,只有大约0.16 eV.体系III 是一个非常值得关注的体系,从能量曲线上可以发现,当分子从电极平面部分转移到表面孤立金原子上时,体系的能量出现约1.2 eV 的大幅度跳跃式下降,这进一步表明了吡啶基在金表面上方适当距离下对附近的表面孤立金原子具有极强的吸引作用.在这一构型变化中,不仅原来与N 原子相连的平面区域的金原子回缩会释放一定量的能量,同时表面吸附的孤立金原子向左移动一个晶格与N 原子相连后也会释放一定量的能量,因此这一过程体系能量下降的幅度明显大于体系I 中分子从第二层金原子转移到尖端金原子时能量下降的幅度.当分子吸附到表面孤立金原子后,随着距离的增加,体系能量在轻微下降后便开始快速上升.更值得关注的是分子离开电极平面区域时的最大作用力只有0.45 nN,但在体系III 中分子从表面孤立金原子上断开却需要高达1.7 nN 的作用力.
以上结果表明,如果4,4′-二吡啶分子一端连接在平面电极上,例如在STM-BJ 技术中,分子连接在基底电极的平面部分,则由于分子与平面电极之间能够承受的拉力太小,分子结在拉伸过程中会直接在分子与平面电极之间断开.而实际用STMBJ 技术组装分子结时,首先将探针电极与基底电极接触,再缓慢提拉探针电极,直到发现电导由1G0突然下降时,说明探针电极刚好与基底电极断开,而后分子以自组装方式连接到两电极之间[2,29,49].由于实验中探针电极需要制作的非常尖锐,在此过程中探针电极很容易从基底电极中拉出孤立的表面金原子.因此在分子形成过程中,分子有极大可能吸附到基底电极表面的孤立金原子上.由于分子与表面孤立金原子可以承受的作用力比较大,足以拉动分子的另一端由探针电极的第二层金原子移动到尖端金原子上,并最后断开,所以在实验中作用力一般会出现由大约1.3—1.5 nN 先下降到0.8—1.0 nN[30,50],而后再完全断开的现象.综上可见,对于4,4′-二吡啶分子结,在实验中通过测量分子结拉伸过程中作用力的变化,可以很好地判断分子结界面的结构演化过程并对关键界面构型进行识别.
虽然体系I 和体系II 的结构演化过程非常相似,而且分子从第二层金原子上断开并转移到尖端金原子上所需要的拉力均大于从尖端金原子上断开所需的拉力.但值得注意的是体系II 中所需的作用力均大于体系I 中相应的作用力,计算发现,这与体系轨道分布以及N-Au 间的成键情况有一定关系.为了直观的理解这一差别,图4 对体系I 和体系II 中同时离域到分子和电极上的占据分子轨道和对N—Au 键有贡献的分子轨道进行了统计,并展示了部分扩展分子轨道的空间分布图,其中对N—Au 键有贡献的分子轨道是指波函数在N—Au 之间没有节面的分子轨道.统计发现对于体系I 和体系II,分子连接到第二层金原子上的离域轨道数目分别为15 条和18 条,对N—Au 键有贡献的轨道数目均为6 条,而当分子连接到尖端金原子上时离域轨道数目分别为10 条和11 条,对N—Au键有贡献的轨道数目均为4 条.因此不管从扩展轨道的数目上,还是从对N—Au 键有贡献的轨道数目上,都显示出分子连接到第二层金原子上时与电极耦合更强,因此分子从第二层金原子上断开需要更大的拉力.另外,这一现象同时也表明分子连接到第二层金原子上与电极之间交换电子的概率更大,这与实验测得分子结在较短电极距离下电导更高是一致的[29,30,49].虽然在相同连接方式下,体系I 和体系II 对N—Au 键有贡献的轨道数目一样,但体系II 的离域轨道数略多于体系I 的轨道数,因此相比于体系I,体系II 中分子断开所需的作用力更大.

图4 体系I、体系II 和体系III 中部分同时离域到分子和电极上的占据轨道的空间分布,其中括号中的数字分别为总的离域占据轨道数目和对N-Au 键有贡献的轨道数目Fig.4.The spatical distributions of part delocalized occupied molecular orbitals for system I,system II and system III.These orbitals are both delocalized on the molecule and electrode.The figures in the brakets are the total numbers of the delocalized occupied molecular orbitals and the orbitals which have contributions to N-Au bond.
对于完全不同的锥形电极和平面形电极,由于电极表面电子态差别较大,从扩展轨道的数量上不能很好的解释分子与电极间的相互作用力,但是从对N-Au 键有贡献的轨道数目上仍然可以比较好地反映出分子与电极间的相对耦合强度.如对于体系III 分子从平面电极的表面孤立金原子上断开所需的作用力明显大于从锥形电极第二层金原子上断开所需的作用力,但是体系III-j 的离域轨道数却只有13 条,明显少于体系I 和体系II 中分子连接到锥形电极第二层金原子上的轨道数.然而体系III-j 中对N-Au 键有贡献的分子轨道有7 条,多于体系I-b 和体系II-f 的6 条,可见对N-Au 键有贡献的分子轨道数对于分子与电极间的耦合强度起着决定作用.之所以4,4′-二吡啶分子与平面电极上吸附的孤立金原子耦合更强,是由于孤立金原子下面近邻的三个金原子还与平面内其他近邻原子存在相互作用,因而减弱了对其上的孤立金原子的束缚,从而导致该孤立金原子与吡啶末端的作用增强.这一点可以从分子与电极间达到最大张力时,平面电极表面孤立金原子和锥形电极尖端金原子离下面一层金原子距离的差别上得到解释.计算表明,对于体系I 和体系II,分子连接到尖端金原子上张力达到最大时,尖端金原子与第二层金原子的距离分别为0.222 nm 和0.233 nm,小于块体金中相邻两原子层之间的间距0.235 nm,说明锥形电极尖端的金原子受的束缚作用明显大于块体金中相邻原子之间的作用.而体系III 中分子连接到表面孤立金原子上张力达到最大时,表面孤立金原子到电极平面的距离为0.240 nm,明显大于块体金中相邻金原子层之间的距离,因此平面电极上的孤立金原子受到的束缚明显减弱.
4 结论
基于密度泛函理论计算了4,4′-二吡啶分子与不同构型电极之间的作用过程.结果显示,分子在远离锥形电极过程中在适当距离下容易接近垂直地连接到电极尖端第二层金原子上,同时尖端的金原子在分子作用下偏向一侧.进一步拉动分子,吡啶末端从第二层金原子转移到尖端金原子上.分子从第二层金原子断开需要1.3—1.5 nN 的拉力,但从尖端电极上断开只需要0.8—1.0 nN 的作用力,与实验观测结果完全一致.平面电极对吡啶末端的吸附作用只有不到0.5 nN,但平面电极表面上的孤立金原子对分子具有很强的吸附作用,可以使4,4′-二吡啶分子承受约1.7 nN 的作用力.由于分子与电极间的作用力与界面演化过程存在明显的对应关系,因此利用基于第一性原理的绝热拉伸模拟方法可以很好地揭示分子结界面结构演化过程和识别分子-电极间特有的界面结构.
