厚叶木莲(Manglietia pachyphylla)基因组草图*
2023-03-01甘新军陈焕锦朱韦光熊露桥余恩萍王峥峰徐凤霞曹洪麟
甘新军,宾 粤,陈焕锦,朱韦光,熊露桥,余恩萍,4,王峥峰**,徐凤霞,曹洪麟
(1.广东从化陈禾洞省级自然保护区管理处,广东广州 510950;2.中国科学院华南植物园,广东省应用植物学重点实验室,中国科学院退化生态系统植被恢复与管理重点实验室,广东广州 510650;3.华南国家植物园,广东广州 510650;4.中国科学院大学,北京 100049;5.中国科学院华南植物园,中国科学院植物资源保护与可持续利用重点实验室,广东广州 510650)
木兰科(Magnoliaceae)是最原始的被子植物,其属种丰富,类型多样,是研究被子植物起源和进化的重要类群[1-4]。我国是木兰科起源地和避难所,为木兰科植物分布中心,拥有很多古老、孑遗和特有物种[2,5-7]。我国的木兰科物种现主要分布于我国热带亚热带地区,如云南省、广西壮族自治区、贵州省和湖南省。木兰科中的很多物种树形优美,叶型秀丽、色泽鲜艳,花形态各异、花色丰富明艳且高贵典雅,观赏性强,是优良的绿化树种[8-11]。木兰科物种的枝、叶、花含有挥发性有机物,作为绿化树种可以净化空气,促进人们身心健康[12-15],而且这些挥发性有机物与木兰科产生的其他次生代谢物如生物碱,常被用作中药[16-18]。另外,木兰科的一些种类具有较强的光合能力、固碳能力以及土壤改良作用,可用于人工林改造,实现林业提质增效[19,20];而且一些木兰科植物树干通直,木材材质均匀、细密,是很好的用材树种[ 8,21,22]。同时,木兰科植物最早记载于我国秦汉时代,历经千年,寄托了人们对美好生活的追求和向往,富有深厚的文化沉淀[23]。
厚叶木莲(Manglietiapachyphylla)为木兰科木莲属(Manglietia)的木本植物,为中国特有种,目前零星分布于我国广东省和广西壮族自治区海拔500 m以上的常绿阔叶林中[24,25]。厚叶木莲的显著特征是有光泽的革质厚叶,可作为园林绿化和用材物种[24]。厚叶木莲种群少,个体数量小,结实率低,种子易被动物啃食,自然更新差[25,26],现被列为国家二级重点保护野生植物[24]。目前国内外已开展厚叶木莲群落学[25,26]、花粉形态[27]和光响应生理[28]等方面的研究,但还未有关于厚叶木莲的遗传多样性及其基因组的研究报道。基因组及基于基因组开展的物种遗传多样性研究,可以揭示物种进化过程,从而了解物种的适应性并进一步应用于品种改良[29]。为此,本研究采用二代和三代高通量测序手段,对厚叶木莲基因组进行测序,组装其基因组草图,为今后更好地开展其进化、遗传多样性研究提供参考。
1 材料与方法
1.1 材料
在广东省广州市从化区陈禾洞省级自然保护区内选择1株厚叶木莲成年大树(生长点地理位置为113°55′31.84″E,23°45′1.25″N),其胸高直径(Diameter at Breast Height,DBH)为22.4 cm,采集其无虫咬和病斑的树叶3片。采集的树叶用剪刀剪碎后,用锡箔纸包裹后立刻投入液氮罐中,之后将样品送至武汉未来组生物技术有限公司进行高通量测序。
1.2 方法
测序包括3个方面的内容:一是采用Nanopore PromethION测序平台对厚叶木莲进行三代基因组测序,二是采用MGI DNBSEQ-T7 测序平台对厚叶木莲进行二代基因组测序,三是采用MGI DNBSEQ-T7 测序平台对厚叶木莲进行转录组测序,具体实验流程参考Wang等[30]的研究。针对所得的测序数据,利用不同程序开展数据处理和基因组组装、分析。在数据处理过程中主要使用程序的默认参数,如在程序运行过程中对默认参数进行改动,则会在文中具体说明。
1.2.1 测序数据前处理
本研究利用Sickle v1.33 (https://github.com/najoshi/sickle)对厚叶木莲二代基因组测序数据进行过滤,去除测序数据中碱基质量小于30、片段长度小于80 bp的测序数据。过滤后的二代测序数据用 RECKONER v1.1[31]进行纠错。对于三代基因组测序数据,由于低质量数据在交付前测序公司已过滤,无需再过滤,本研究只利用Porchop 0.2.4 (https://github.com/rrwick/Porechop)对厚叶木莲三代基因组测序数据进行接头过滤。
1.2.2 基因组大小预测与组装
针对厚叶木莲二代测序数据,利用GenomeScope 2.0[32]进行基因组大小预测(参数“-k 21”)。针对厚叶木莲三代测序数据,本研究选择大于10 kb测序序列,利用NextDenovo 2.3.1(https://github.com/Nextomics/NextDenovo)进行基因组组装,组装的基因组利用Pseudohaploid (https://github.com/schatzlab/pseudohaploid)和Purge_Dups v1.2.6[33]去除冗余序列(如杂合导致的拼接序列),之后利用racon v1.5.0[34],hapo-G v1.3.2[35]和polypolish v0.5.0[36]进行组装序列纠错。针对组装完成的基因组,利用BUSCO (Benchmarking Universal Single-Copy Orthologs)v5.4.6[37]对照“eudicots_odb10.2020-09-10”和“embryophyta_odb10.2020-09-10”两个单拷贝基因库进行组装完整性的评估。
1.2.3 重复序列和基因的预测、注释
本研究利用EDTA (Extensive De-novo TE Annotator)v2.1.0[38]和RED (REpeat Detector)v2.0[39]预测厚叶木莲基因组中的重复序列。利用BEDTools v2.29.2[40]中的“merge”命令将两个重复序列预测结果合并,再利用“maskfasta”命令将预测的重复序列屏蔽掉。针对屏蔽了重复序列的基因组,首先利用BRAKER2[41],同时结合转录组数据和9个物种的蛋白质序列(表1)预测厚叶木莲基因组组装序列中的基因;然后将结果输入funannotate pipeline v1.8.13(https://github.com/nextgenusfs/funannotate)中,同样结合转录组数据和9个物种的蛋白质序列,进一步预测厚叶木莲基因组组装序列中的基因,这一过程包括3个步骤和命令:“funannotate train”“funannotate predict”和“funannotate update”,在“predict”步骤中使用的参数为“-max_intronlen 100,000 -busco_db embryophyta -organism other”。
表1 预测厚叶木莲基因组基因的参考物种
基因预测结束后,利用8个不同的蛋白质注释平台对预测的基因进行功能分析,包括:dbCAN (DataBase for automated Carbohydrate-active enzyme ANnotation)v10.0[42],eggNOG-mapper (Evolutionary genealogy of genes:Non-supervised Orthologous Groups-mapper)v5.0.2[43],GO (Gene Ontology)[44,45],KEGG (Kyoto Encyclopedia of Genes and Genomes)[46],InterPro (The Integrated Resource of Protein Domains and Functional Sites)v5.60-92.0[47],MEROPS v12.0[48],Pfam (The protein families database)v35.0[49]和SignalP 5.0b[50]。
考虑到注释的基因中有可变剪切的状况,在进行比较基因组的研究中去除了各物种基因中的可变剪切产生的基因,只保留其中最长的基因序列进行分析。
1.2.4 基因家族和系统发育分析
利用OrthoFinder 3.0.0[51,52]并结合其他9个物种(表1)的蛋白质序列进行基因家族分析。在OrthoFinder分析过程中,程序自动分析获得物种间的单拷贝基因,并用这些单拷贝基因进行系统发育树构建。利用构建的系统发育树,使用MCMCTree[53]进行物种间分化时间的估算。在这一分析过程中需要参考已有物种间的分化时间,本研究从http://timetree.org/获得这些信息,并在表2中列出。在得到有分化时间的系统发育树后,再利用cafe (Computational Analysis of gene Family Evolution)v5[54]进行基因家族的扩张和收缩分析;对其中显著扩张和收缩的基因家族,则利用 TBtools v1.115[55]进行GO和KEGG的富集分析。
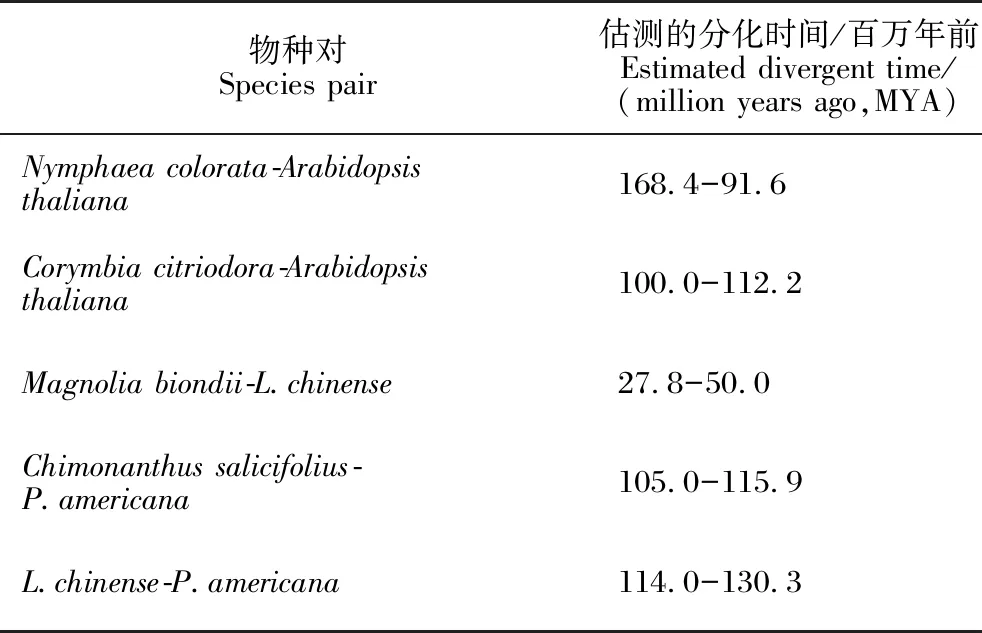
表2 从http://timetree.org/获得的物种对间分化时间
1.2.5 基因重复(Gene duplications)
利用wgd v1.1.2[56]进行基因组的全基因组重复(Whole genome duplication)事件分析,该过程是在基因组中进行基因间的同源性分析,找到两两同源基因,并对两两同源基因进行同义突变率(synonymous substitution rate,Ks)计算,之后查看Ks值的密度分布图,其峰值出现的地方提示有全基因重复事件发生;进一步利用 Doubletrouble v0.99.1 (https://github.com/almeidasilvaf/doubletrouble)开展全基因组重复基因、串联重复(Tandem duplications)基因、近端重复(Proximal duplications)基因、转座重复(Transposed duplications)基因、散在重复(Dispersed duplications)基因的分析[55]。其中,串联重复基因是指彼此连续或中间间隔不超过5个其他基因的近缘基因;近端重复基因是连续分布、中间间隔不超过10个其他基因的近缘基因;转置重复基因是由转座子介导的重复基因;散在重复基因是随机分布、彼此间不靠近的重复基因[57]。利用TBtools分别对全基因组重复基因、串联重复基因、近端重复基因进行GO和KEGG富集分析。
2 结果与分析
2.1 厚叶木莲基因组测序与组装
本研究中三代基因组测序共获得大约118.1 Gb测序数据,二代基因组测序获得约264.1 Gb测序数据,转录组测序获得约31.4 Gb测序数据。基因组和转录组原始测序数据已上传至GenBank,三代基因组测序数据序列号为SRR24423593、SRR24423594、SRR24423595;二代基因组测序数据序列号为SRR24390471、SRR24390472、SRR24390473;转录组测序数据序列号为SRR24415003。利用GenomeScope 2.0分析厚叶木莲基因组大小为1 969 269 649 bp。
本研究利用NextDenovo 2.3.1进行厚叶木莲基因组组装,得到基因组组装大小为2 350 821 062 bp,包含1 436个拼接序列(contig),N50(将组装的序列按照长度由大到小进行累加,当累加到某个序列时,累加的值为基因组50%的长度时,此序列的长度即为N50)为6 839 193 bp,最长拼接序列为26 073 671 bp,最短拼接序列为16 675 bp,平均为1 637 062.0 bp。去除冗余序列和纠错后,最终的基因组组装大小为2 092 298 891 bp,包含676个拼接序列,N50为7 961 115 bp,最长拼接序列为26 180 362 bp,最短拼接序列为27 281 bp,平均为3 095 117 bp,组装的其他统计信息见表3。组装的厚叶木莲基因组数据上传至GenBank,序列号为JASAUF000000000。
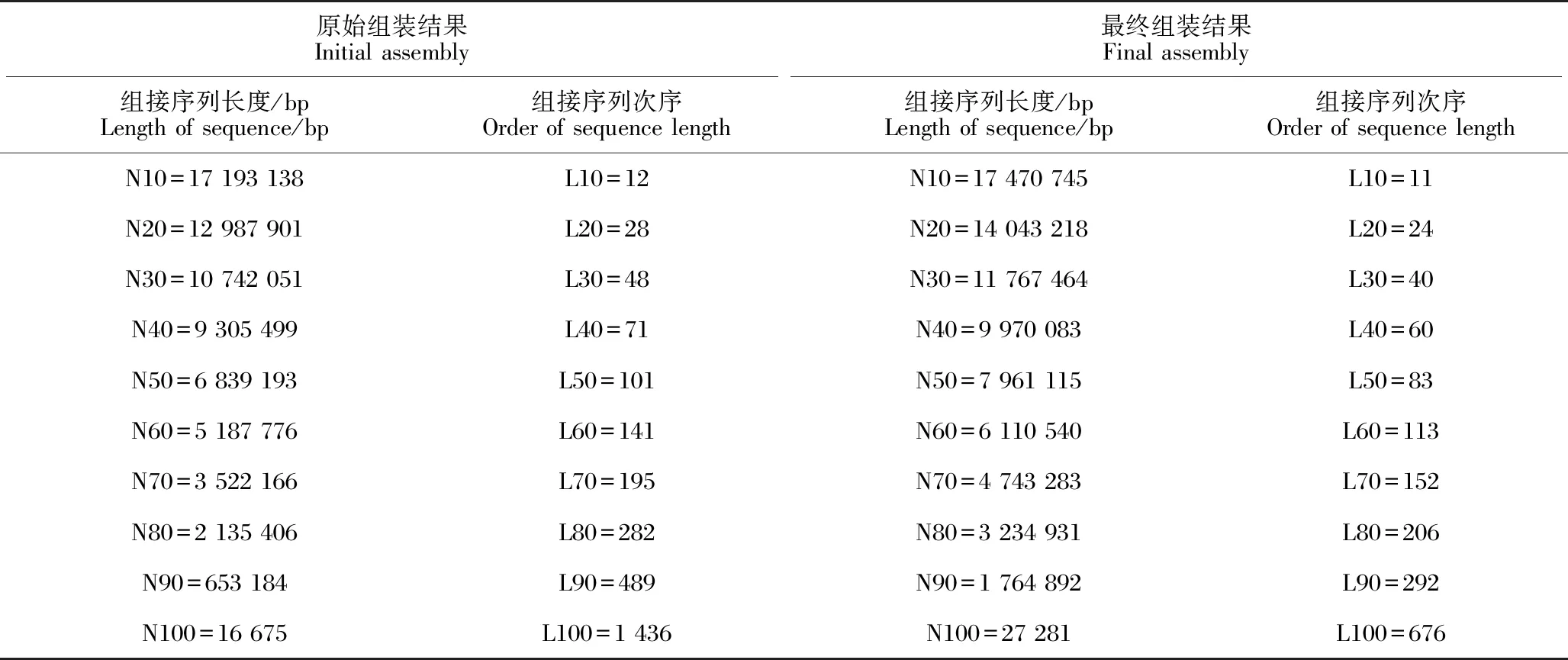
表3 厚叶木莲基因组组装结果
BUSCO对组装的基因组完整性的评估显示,比对“eudicots_odb10.2020-09-10”单拷贝基因库,全部2 326个BUSCO单拷贝基因,96.6% BUSCO单拷贝基因在厚叶木莲基因组中完整匹配。其中,能完整匹配到BUSCO单拷贝基因库而且在厚叶木莲基因组中也是单拷贝的基因占90.8%,能完整匹配到BUSCO单拷贝基因库但在厚叶木莲基因组中为多拷贝的基因占5.8%;不完整匹配到BUSCO单拷贝基因库的基因有27个,占1.2%;有54个BUSCO单拷贝基因未在厚叶木莲基因组的基因中匹配到,占2.3%。比对“embryophyta_odb10.2020-09-10”单拷贝基因库,全部1 614个BUSCO单拷贝基因中,98.8% BUSCO单拷贝基因在厚叶木莲基因组中完整匹配,能完整匹配到BUSCO单拷贝基因库而且在厚叶木莲基因组中也是单拷贝的基因占95.3%,能完整匹配到BUSCO单拷贝基因库但在厚叶木莲基因组中为多拷贝的基因占3.5%;不完整匹配到BUSCO单拷贝基因库的基因有12个,占0.7%;有7个BUSCO单拷贝基因未在厚叶木莲基因组的基因中匹配到,占0.4%。
2.2 厚叶木莲基因组注释
在厚叶木莲基因组中,RED和EDTA分别检测到1 342 604 397 bp (64.16%)和1 447 436 839 bp(69.17%)重复序列,两者合并后得到1 601 108 919 bp的重复序列,占基因组的76.52%。EDTA检测结果表明,厚叶木莲基因组中重复序列最多的是Gypsy类的长末端重复序列(Long terminal repeat),有475 508 695 bp,占基因组的22.73%(表4)。
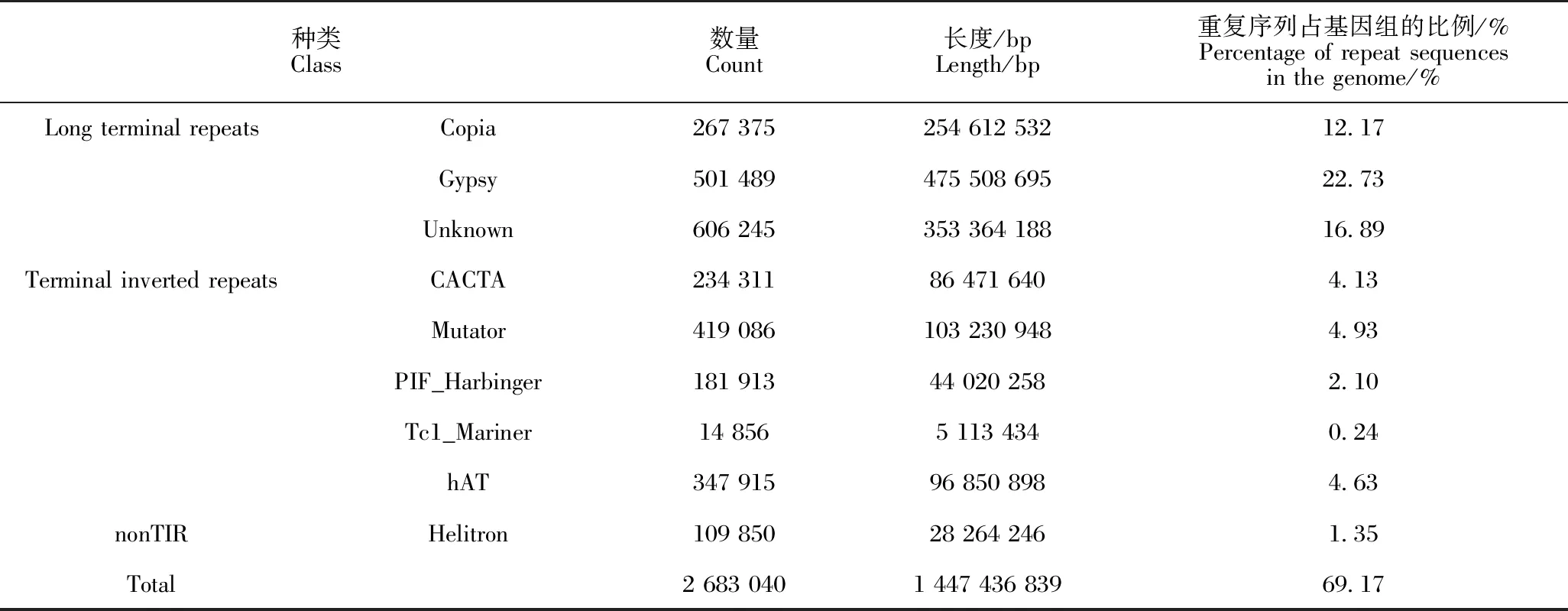
表4 EDTA检测的重复序列
本研究对厚叶木莲基因组进行基因预测共获得37 900个基因,这些基因共编码了41 675种蛋白质。利用数据库对这些蛋白质序列进行功能注释,其中32 249个 (77.4%)蛋白质序列注释到了8个蛋白质注释平台数据库中的一个,具体如下:17 886个蛋白质序列注释到GO数据库,21 430个蛋白质序列注释到InterPro数据库,31 146个蛋白质序列注释到eggNOG-mapper数据库,23 203个蛋白质序列注释到Pfam数据库,1 160个蛋白质序列注释到dbCAN数据库,1 016个蛋白质序列注释到MEROPS数据库,3 078个蛋白质序列注释到SignalP数据库,15 752个蛋白质序列注释到KEGG数据库。基因注释文件已上传至中国科学院华南植物园数据存储库(https://doi.org/10.57841/casdc.0001214)。
2.3 厚叶木莲基因家族与基因组进化分析
本研究对包括厚叶木莲在内的10个物种的蛋白质序列进行了基因家族分析,结果共得到24 616个基因家族。从表5统计结果可以看出,厚叶木莲基因组中有34 319个基因归为其中一个基因家族,占总基因的90.6%。对于木兰科其他两个物种鹅掌楸和望春玉兰,它们分配到基因家族的基因比例分别为95.7%和93.4%,均高于厚叶木莲;但厚叶木莲基因分布于15 894个基因家族中,占全部基因家族的64.6%;而鹅掌楸和望春玉兰的基因出现在13 858和14 935个基因家族中,占全部基因家族的56.3%和60.7%,低于厚叶木莲。24 616个基因家族中有710个厚叶木莲特有的基因家族,这些家族包含了3 373个厚叶木莲基因。鹅掌楸和望春玉兰特有的基因家族分别是437和999个,虽然一个低于厚叶木莲特有基因家族数目,一个高于厚叶木莲特有基因家族数目,但是它们所包含的基因数(分别为6 845和4 543)均高于厚叶木莲,而且这些特有基因数占两个物种各自所有基因的比例也高于厚叶木莲,特别是鹅掌楸,为19.4%(表5,图1)。3种木兰科植物共同出现的基因家族有345个(图1)。

图1 厚叶木莲与其他物种基因家族交集图
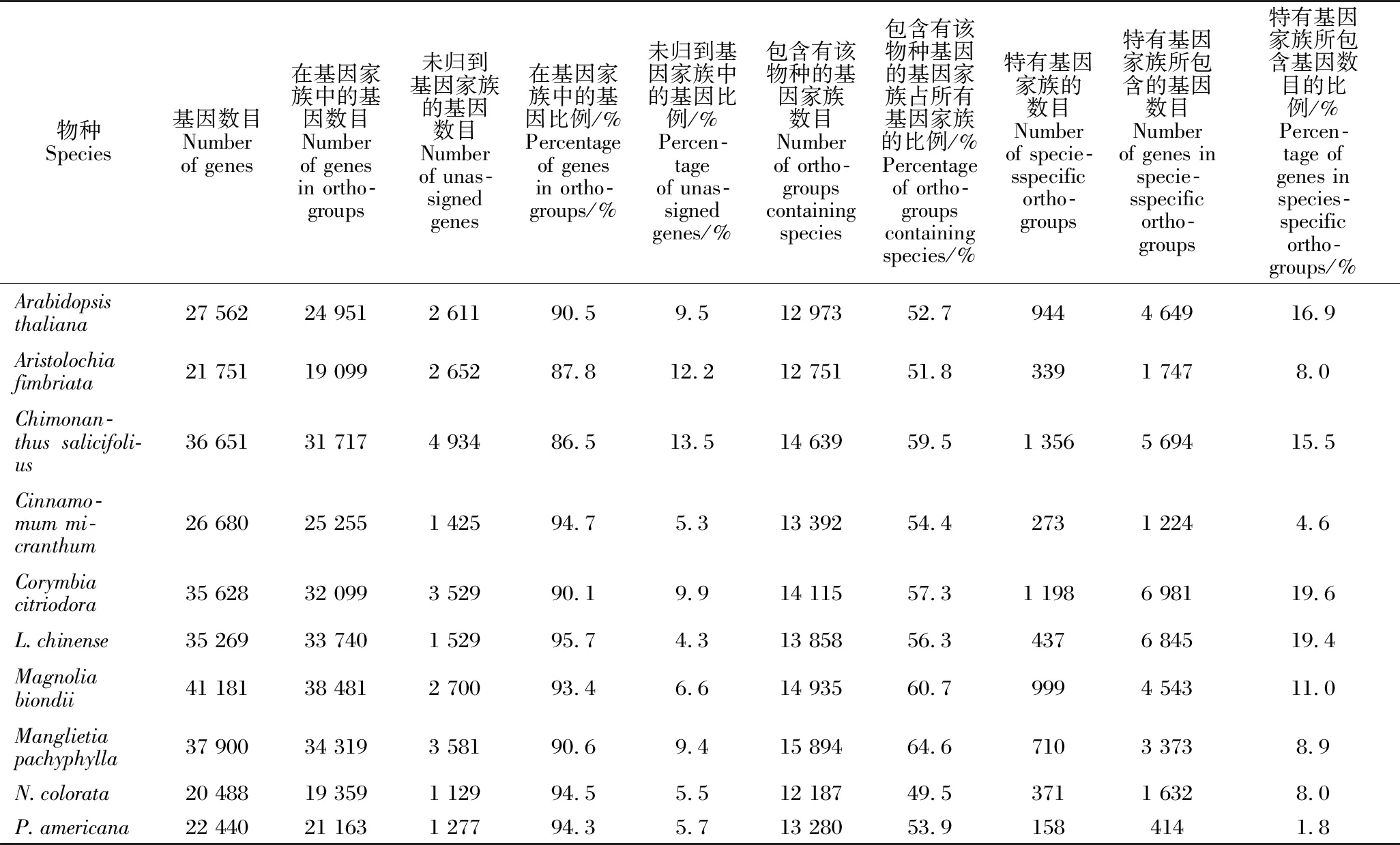
表5 基因家族及其基因信息统计
对厚叶木莲特有的基因家族进行GO和KEGG富集分析,结果表明这些基因家族所包含的基因的主要功能与细胞内稳态(GO:0055067、GO:0006885、GO:0030433、GO:0036503)、葡聚糖代谢(Glucan metabolic process)(GO:0044042、GO:0016762、GO:0046527)、原核生物抗性(Prokaryotic defense system)、苯丙氨酸代谢(Phenylalanine metabolism)和转录(Transcription)等有关(https://doi.org/10.6084/m9.figshare.23690001.v3,表S1和S2)。
系统发育分析表明厚叶木莲与望春玉兰聚在一起(图2),两个物种是在约10 500 000年前(95%CI:4 989 810-17 055 600年)从共同的祖先开始分化。对基因家族扩张和收缩分析结果表明,厚叶木莲基因组中有686个基因家族表现为收缩,1 295个基因家族表现为扩张,其中有136个基因家族表现为显著收缩,417个基因家族表现为显著扩张。GO和KEGG富集分析表明,厚叶木莲基因组中显著扩张的基因家族与木质部、韧皮部发育(GO:0010088,GO:0010087)、肌动蛋白丝(Actin filament,GO:0030837、GO:0030833、GO:0061572、GO:0051017、GO:0030832、GO:0008064、GO:0030041)、细胞运动(Cell motility)、油菜素甾醇生物合成(Brassinosteroid biosynthesis)、类黄酮生物合成(Isoflavonoid biosynthesis)、维生素B6代谢(Vitamin B6 metabolism)、硫代葡萄糖苷(Glucosinolate biosynthesis)和萜类物质生物合成有关(https://doi.org/10.6084/m9.figshare.23690001.v3,表S3和表S4);厚叶木莲基因组中显著收缩的基因家族主要与木质素(Lignin)代谢(GO:0009808、GO:0046274)、类黄酮生物合成,以及二苯乙烯、二芳基庚酸类和姜酚生物合成(Stilbenoid,diarylheptanoid and gingerol biosynthesis)相关(https://doi.org/10.6084/m9.figshare.23690001.v3,表S5和表S6)。
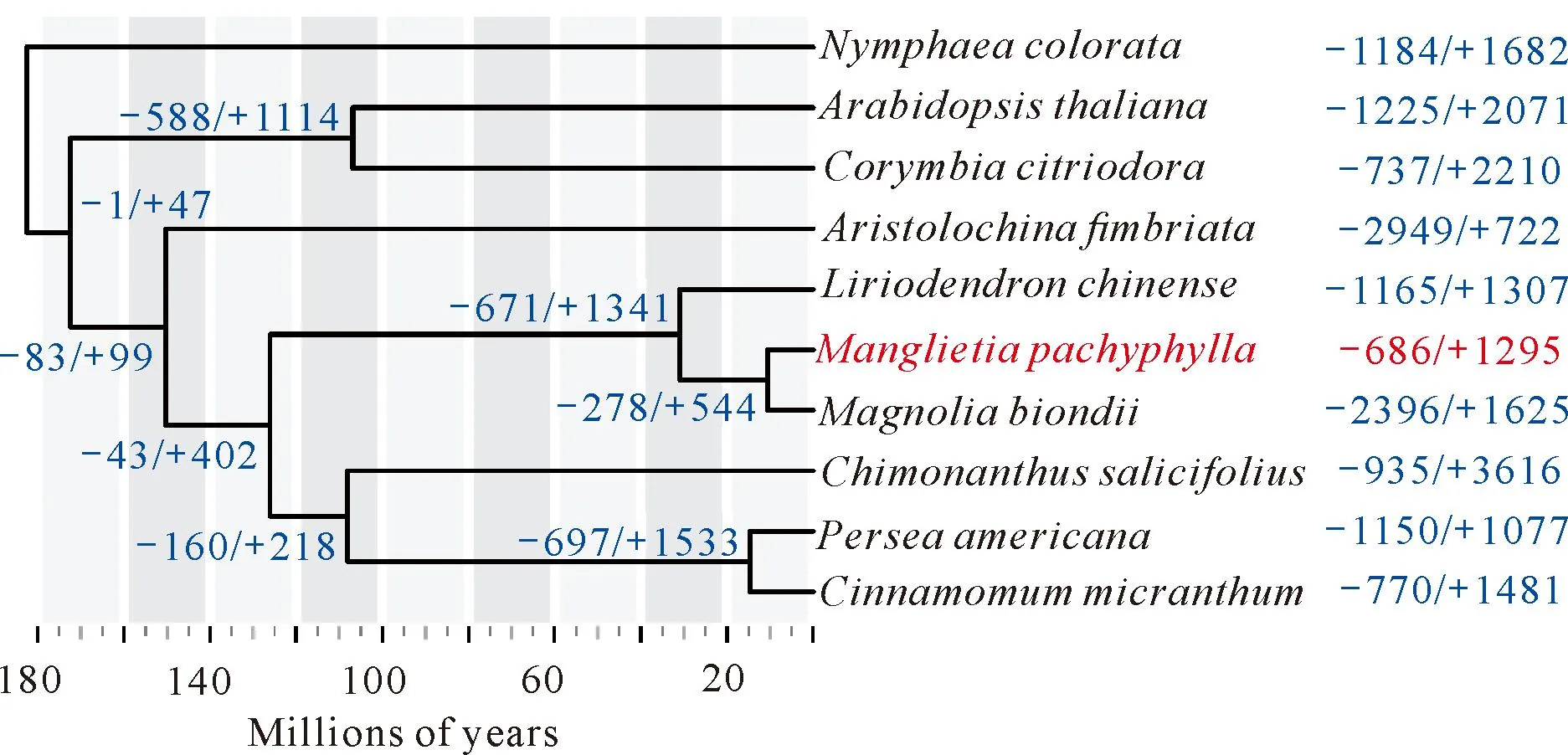
Divergence times are shown below the tree;The "-/+" and the numbers beside the tree nodes and species represent the number of contracted and expanded gene families in Manglietia pachyphylla and other species.
2.4 厚叶木莲全基因组和基因重复分析
全基因组重复事件分析表明,厚叶木莲和鹅掌楸、望春玉兰表现出同样的Ks峰型(图3),因此它们近期共同经历了一次全基因组重复事件。对厚叶木莲基因组的基因重复研究表明,厚叶木莲基因组中有4 769个基因与其全基因组重复有关(12.6%),3 317个基因为串联重复(8.8%),3 124个基因为近端重复(8.2%),136个基因为转置重复(0.4%),18 522个基因为散在重复(48.9%)。GO和KEGG富集分析表明,厚叶木莲基因组中全基因组重复基因主要功能与DNA内复制调控(Regulation of DNA endoreduplication)、植物昼夜节律(Circadian rhythm-plant)、鸟嘌呤核苷酸结合蛋白(GTP-binding proteins)、柠檬酸循环[Citrate cycle (TCA cycle)],蛋白酶体(Proteasome)、转录因子(Transcription factors)(https://doi.org/10.6084/m9.figshare.23690001.v3,表S7和表S8)相关。GO和KEGG富集分析表明,厚叶木莲基因组中串联重复基因与NADH氧化(NADH oxidation,GO:0006116)、寡肽转运(Oligopeptide transport,GO:0006857)、RNA脱帽(RNA decapping,GO:0110154)、谷胱甘肽代谢过程(Glutathione metabolic process,GO:0006749)、过氧化氢分解代谢过程(Hydrogen peroxide catabolic process,GO:0042743、GO:0042744)、类黄酮生物合成、牛磺酸和低牛磺酸代谢(Taurine and hypotaurine metabolism)、苯并恶唑嗪酮类化合物生物合成(Benzoxazinoid biosynthesis)、玉米素生物合成(Zeatin biosynthesis)、萜类生物合成(Terpenoid biosynthesis)、苯丙烷类生物合成(Phenylpropanoid biosynthesis)、生物碱合成等相关(https://doi.org/10.6084/m9.figshare.23690001.v3,表S9和表S10)。厚叶木莲基因组中近端重复基因主要和防御(GO:0043207、GO:0098542、GO:0051707、GO:0009607、GO:0050832、GO:0009605、GO:0006952)、次生代谢(GO:0044550、GO:0019748)、各类生物碱生物合成(Biosynthesis of various alkaloids)、花青素生物合成(Anthocyanin biosynthesis)、聚酮生物合成(Polyketide biosynthesis)、苯并恶唑嗪酮类化合物生物合成、萜类生物合成、硫代葡萄糖苷生物合成和油菜素甾醇生物合成等相关(https://doi.org/10.6084/m9.figshare.23690001.v3,表S11和表S12)。

图3 全基因组重复分析中同义突变率(Ks)的密度分布
3 讨论
3.1 厚叶木莲的基因组组装
高通量测序已成为研究物种基因组和遗传多样性的主要手段。目前高通量测序分为短片段测序和长片段测序量两种方式,前者测序平台主要包括Illumia和MGI测序公司一系列仪器设备,后者测序平台主要包括Nanopore和PacBio测序公司一系列仪器设备。短片段高通量测序的优势是测序的准确性较高、数据量大和价格便宜;长片段测序的优势是测得的序列长度长、完整性好,有利于后续基因组组装的连续性,如Nanopore测序平台可测100 kb或更长的序列片段。但采用Nanopore和PacBio测序平台测得的序列错误率较高,为此PacBio公司推出了采用Hifi (High fidelity reads)模式的测序方式,使得测序错误率大为降低,但是测序长度有所限制(15-20 kb)。为得到高质量基因组,可采用不同测序平台测序,再进行基因组组装,以实现优势互补。本研究采用Nanopore长片段测序平台的测序结果进行厚叶木莲的基因组初步组装,再用MGI短片段测序平台的测序结果对组装的基因组进行纠错,得到了较完整和准确的组装结果,但是还没有组装到染色体级别,后期本研究将继续采用Hi-C(High-throughput chromosome conformation capture)测序方式进一步对厚叶木莲基因组进行测序,测得的结果可用于进一步组装以完善厚叶木莲基因组。
在采用Nanopore测序数据进行厚叶木莲基因组组装过程中,本研究采用NextDenovo 2.3.1软件进行序列拼接,这一软件采用先纠错再组装(Correction then assembly)的模式进行基因组组装,保证了组装序列的连续性和正确性,优于其他组装软件。同时本研究利用多种方法去除了原始组装序列中的冗余序列并进行后期纠错:其中racon v1.5.0和hapo-G v1.3.2分别使用Nanopore测序获得的长片段和MGI测序获得的短片段进行组装基因组内碱基和插入缺失错误的纠错,polypolish v0.5.0主要针对组装序列中的同聚体长度(Polypolish-length,如“AAAAAA”这种重复序列的长度)进行纠错,3个纠错软件的使用可以很好地保证组装基因组的准确性。
在基因组注释环节,本研究利用EDTA和RED软件相互组合的方法进行基因组中重复序列的查找。EDTA主要用于基因组中转座子的查找,而RED查找包括转座子在内的所有重复序列类型,如微卫星体(Microsatellite)等,两者结合保证了重复序列查找的全面性,但RED分析的结果并没有对不同重复序列进行归类。本研究首先使用BRAKER2,结合转录组数据和其他物种的蛋白质序列预测厚叶木莲基因组中的基因,得到初步基因序列注释结果。由于这一结果还存在较多错误预测,本研究进一步利用funannotate软件对初步预测的结果进行整合。Funannotate是个软件整合工具,被编译为一个管段(Pipeline)流程。该软件可以进行不同方式的基因预测(包括de novo注释、转录组注释和同源蛋白质注释),再结合其他分析软件的结果(本研究中为BRAKER2基因预测结果),可以获得统一、可靠的高质量基因预测结果。
3.2 厚叶木莲的比较基因组学研究
本研究中厚叶木莲的基因组组装大小为2 092 298 891 bp(约2.09 Gb),大于木兰科的日本厚朴(Magnoliahypoleuca,修正名为Houpoeaobovata)基因组(约1.64 Gb)[58]、厚朴(Magnoliaofficinalis,修正名为H.officinalis)的基因组(约1.68 Gb)[59]和鹅掌楸的基因组(约1.74 Gb)[60],小于望春玉兰的基因组(约2.22 Gb)[29]。基于“embryophyta_odb10.2020-09-10”单拷贝基因库,厚朴基因组的组装完整性评估为86.20%的BUSCO单拷贝基因能被完整匹配到,日本厚朴基因组的组装完整性为98.6%,望春玉兰基因组的组装完整性为95.7%(上述数值为本研究利用望春玉兰基因组分析的结果),鹅掌楸基因组的组装完整性为98.8%(此值为本研究利用鹅掌楸基因组重新分析的结果),而厚叶木莲的BUSCO组装完整性为98.8%,与鹅掌楸的结果相同,但高于厚朴、日本厚朴和望春玉兰3个物种。从重复序列比例看,厚叶木莲基因组中重复序列占基因组的76.5%,高于望春玉兰(66.48%)[29]、日本厚朴(64.54%)[58]以及鹅掌楸(63.81%)[60],但小于厚朴(81.44%)[57],厚叶木莲基因组重复序列比例在已有木兰科物种基因组重复序列比例的范围内。上述结果说明,虽然本研究报道的厚叶木莲基因组还是草图状态,没有组装到染色体级别,但是组装已经很完整,结果可靠。
对基因家族的研究表明,厚叶木莲中与次生代谢相关的基因家族显著扩张,如萜类物质,这与日本厚朴、望春玉兰基因家族分析结果相似[29,58]。基因表达分析表明,望春玉兰花中萜类相关基因的表达高于其叶子,说明萜类物质是望春玉兰花香的主要原因[29],厚叶木莲花是否也有类似的结果还需结合基因组进一步研究确定。除了次生代谢物质相关的基因家族在厚叶木莲基因组中有显著扩张外,本研究还发现与木质部、韧皮部发育相关的基因家族,以及与光合作用[光合作用光反应(Photosynthesis light reaction,GO:0019684)、光合作用光收获(Photosynthesis light harvesting,GO:0009765)以及KEGG中的光蛋白(Photosynthesis proteins)]、温度[热反应(Response to heat,GO:0009408)、温度刺激反应(Response to temperature stimulus,GO:0009266)]等相关的基因家族也有扩张。厚叶木莲生长在较高海拔的地区,木材致密通直[26],这些扩张的基因家族是否与其适应高海拔的温度、光照、强风等生境有关还需进一步研究。厚叶木莲基因组中的扩张基因家族中的一部分与肌动蛋白丝(也称“微丝”)相关,而肌动蛋白是植物细胞骨架的主要元素,相关研究表明它与植物抗真菌功能密切相关[61,62]。
对日本厚朴、望春玉兰的研究均表明木兰科都共同经历了一次近期的全基因组重复事件[29,58]。厚叶木莲有着与日本厚朴相似的全基因组重复基因、串联重复基因和近端重复基因比例。与全基因组重复相关的基因占厚叶木莲基因组基因的比例为12.6%,日本厚朴为13.4%[58];串联重复基因在厚叶木莲基因组基因的比例为8.8%,日本厚朴为7.6%[58];近端重复基因在厚叶木莲基因组基因的比例为8.2%,日本厚朴为9.4%[58]。基因串联重复和近端重复是基因形成的重要方式,与物种适应性密切相关[57]。日本厚朴中串联重复和近端重复基因的功能主要与苯丙烷类、萜类、类黄酮的生物合成等有关[58],这与厚叶木莲这两类基因的研究结果相同,但厚叶木莲可产生更多的次生代谢合成物,如苯并恶唑嗪酮类化合物、生物碱、聚酮等。厚朴基因组中,萜类合成相关的基因也呈串联重复状况[59]。值得注意的是日本厚朴原产日本,具有很好的抗寒性,研究发现苯丙烷类生物合成与日本厚朴抗寒性相关。
对厚叶木莲中与全基因组重复相关基因的富集分析表明,这些基因与植物最基本的生长发育密切相关,如植物昼夜节律相关基因[63]、与碳代谢相关的柠檬酸循环基因[64]、参与细胞的多种生命活动(如细胞通讯、核糖体与内质网的结合、小泡运输、蛋白质合成等)的GTP结合蛋白基因[65,66]等,说明全基因组重复在木兰科植物适应性方面具有重要作用。
4 结论
本研究首次报道了木兰科木莲属物种的基因组,这一结果对全面深入了解木兰科物种以及厚叶木莲的进化、适应具有重要作用。本研究组装的厚叶木莲基因组大小为2 092 298 891 bp,基因组序列中76.5%为重复序列。通过基因预测,在组装的厚叶木莲基因组中共注释到37 900个基因,它们编码了41 675种蛋白质。对厚叶木莲基因组研究发现,全基因组重复与木兰科植物进化适应性密切相关,很多与植物基本生长发育相关的基因通过全基因组重复在木兰科得到加强;厚叶木莲富含有与次生代谢相关的多种基因,由此产生的次生代谢物质有利于厚叶木莲在高海拔生长以及抵御病虫害,但其(种子)香味也使得其易受啃食伤害。因此,厚叶木莲基因组的组装为从遗传角度深入了解其濒危机制提供了可能,也为科学合理利用厚叶木莲以及提取其次生物质作为生物医药提供了参考。
