钒钛系脱硝催化剂抗硫酸氢铵中毒改进措施研究进展
2023-03-01刘亮王朝曦李鑫龙张高山王守阳张林林陆畅卿梦霞
刘亮,王朝曦,李鑫龙,张高山,王守阳,张林林,陆畅,卿梦霞
(1 长沙理工大学能源与动力工程学院,湖南 长沙 410114;2 中国华电集团有限公司湖南分公司,湖南 长沙410000)
氮氧化物(NOx)的排放会导致酸雨、光化学烟雾、臭氧消耗和水体富营养化等危害,是最主要的大气污染物之一[1−3]。近些年来燃煤烟气超低排放政策得到广泛关注,2021 年3 月,“十四五”规划和2035 年远景目标纲要中提出推进煤电灵活性改造要求,并强调进一步降低NOx等污染物排放。利用NH3作为还原剂的选择性催化还原工艺(NH3−SCR)是我国燃煤电厂等固定源烟气中NOx脱除的主流技术,其主要反应过程见式(1)、式(2)[4−5]。
脱硝催化剂是SCR技术的核心部分,其中V2O5/TiO2催化剂因在中温段(300~450℃)具有较高的脱硝活性和抗硫性而被广泛应用。但当锅炉运行负荷较低时,SCR 脱硝装置入口烟气温度较低,V2O5/TiO2催化剂的活性下降,因而燃煤电厂中常设置有最低连续喷氨温度,导致这一现象的主要原因是SCR 反应过程中NH3会与烟气中的SO3反应生成相应的硫酸盐物质。其中硫酸铵(AS)可以通过吹灰去除,且受热易分解,对催化剂活性影响不大;而硫酸氢铵(ABS)具有黏性,吹灰很难去除,其热分解温度为400℃左右,因此导致低温下ABS 会在催化剂表面大量沉积导致催化剂失活[6]。此外,脱硝反应过程中常伴随SO2向SO3催化氧化的副反应,进一步促进了催化剂表面ABS的沉积[7]。
综上所述,ABS在催化剂表面沉积导致其失活是低温SCR催化剂研发中的关键问题之一,全面了解ABS 在钒钛系催化剂表面的沉积/分解行为对保障SCR脱硝系统长期稳定运行以及开发和制备新型低温SCR催化剂具有重要意义。基于此,本文分析了钒钨钛系催化剂表面ABS的生成、沉积及演化机理,并重点讨论了现阶段抗ABS中毒的新型钒钛系商业低温催化剂的研发问题,为后续低温抗ABS中毒的SCR脱硝催化剂的研究提供一定的理论指导。
1 ABS生成机理
烟气中ABS 生成机理主要有两种:气相反应机理和催化剂表面反应机理。其中气相反应机理又分为气相一步反应机理和气相两步反应机理。
(1)气相一步反应机理[8]未反应的NH3直接与烟气中的SO3和H2O发生反应生成ABS,见式(3)。
(2)气相两步反应机理[9]①烟气中的SO3与H2O 发生反应生成H2SO4;②H2SO4与NH3发生反应生成ABS,见式(4)、式(5)。
在ABS 的气相反应机理中,H2SO4浓度决定气相反应中式(3)和式(5)的主导地位,有学者[10]应用热力学内容对式(4)的化学平衡常数K进行了研究,见式(6)。
发现当反应温度低于300℃时,SO3主要以H2SO4的形式存在,ABS 生成反应主要按气相两步反应机理进行。
钒钛系催化剂表面反应机理[6,11−12]为,钒钛系催化剂表面ABS 有两种生成途径。一是配位结合催化剂表面V= = O基团中Lewis酸活性位点的NH3,与烟气中的O2、SO2发生反应生成ABS,见式(7)。
二是SO2与催化剂表面的V2O5反应生成的吸附态金属氧化物硫酸盐(VOSO4物种)与气态NH3直接反应生成ABS,见式(8)、式(9)。
烟气中ABS 的生成与反应物的体积分数密切相关,其中ABS 的生成量与生成温度均随反应物体积分数的增大而升高。实际条件下250~350℃烟气温度范围内SCR 反应过程中催化剂表面均会有ABS 的生成[13−16],而喷氨量过多会增加硫酸盐以及硫酸氢盐细颗粒物的浓度[17]。SCR 脱硝过程中燃煤烟气组分复杂,除烟气中SO3与NH3的体积分数直接影响ABS 生成外,其他气体组分也将通过改变相应SO3或NH3的浓度而间接影响ABS 的生成。其中烟气中的NOx对ABS的生成起促进作用,烟气中的NO2能直接在气相中将SO2氧化成SO3[18],见式(10)。
而NO 则可通过促进催化剂表面低价钒向高价钒的转化而促进催化剂表面SO2的催化氧化[19−20]。此外,NH3浓度的增加也常伴随着SO3生成率的增加,进一步促进烟气中ABS的生成[17]。
烟气中相应气体组分除影响ABS 的生成量与生成温度外,NH3/SO3摩尔比的不同也将直接影响相应沉积物的生成。在高温(223~390℃)条件下生成物均为液态ABS,而低温(131~223℃)条件下,当NH3/SO3比为1∶2时,生成物为以密集小液滴形式沉积的H2O、H2SO4与少量ABS 的混合物;当1<NH3/SO3<2 时,生成物中ABS 和AS 同时存在,当NH3/SO3比为2∶1时,生成物为以白色雾状形式沉积的AS[21−22]。
2 ABS在催化剂表面的沉积
燃煤电厂SCR 反应温度在300~420℃之间,常高于ABS结露温度(290℃[23]),但实际条件下仍存在催化剂ABS 堵塞现象,这主要是由ABS 在催化剂孔道结构中的毛细冷凝现象导致的[8,10,23]。毛细冷凝现象是指当气体在低于饱和蒸气压时遇到毛细孔、多孔等结构发生的液化凝聚现象,即物质由气相凝结成液相并存在于毛细结构或多孔介质的孔洞中[12,24]。钒钛系脱硝催化剂具有大量的微孔和介孔,当孔隙结构内的气压小于ABS 的饱和蒸气压时,ABS由气态凝结成液态。根据Thomson毛细凝聚理论[10],半径为r的微孔中蒸气压可通过式(11)计算。
式中,peq为ABS 的平衡蒸气压,Pa;p为ABS在微孔中的蒸气压,Pa;σ为表面张力,N/m;rpore为微孔半径,m;M为ABS的摩尔质量,kg/mol;ρ为ABS的密度,kg/m3;R为气体常数;T为热力学温度,K。由式(11)可知,当ABS 在微孔中的蒸气压大于理论值时就会结露。SCR运行过程中ABS主要以液体形式沉积于催化剂狭窄孔隙中,低温且催化剂孔径较小时,更容易出现ABS 沉积、堵塞的现象。为避免ABS 在催化剂孔隙内的沉积与堵塞问题,实际燃煤电厂中SCR 的最低连续运行温度常在290℃以上。
ABS 在催化剂表面沉积后,会导致其活性降低。一方面,ABS的沉积会堵塞催化剂的孔道、覆盖催化剂表面的活性位点,影响反应物在催化剂中的扩散与吸附,导致催化剂活性降低[25]。研究发现,与未负载ABS 的催化剂相比,表面负载ABS后其比表面积和孔体积均降低了50%左右[26]。如图1 所示,催化剂表面ABS 负载量在1%~5%时孔径分布与未负载ABS 样品基本相同,小孔数量有所减少;ABS负载量增加到10%后,催化剂全部尺寸孔的数量均大量减少;当负载量到达30%时,ABS完全覆盖催化剂表面,内部孔隙结构几乎完全被堵塞,催化剂严重失活[27]。另一方面,ABS沉积在催化剂表面后,也会与催化剂存在相互作用,显著降低催化剂中V5+的含量,从而降低催化剂的氧化还原性能[28]。
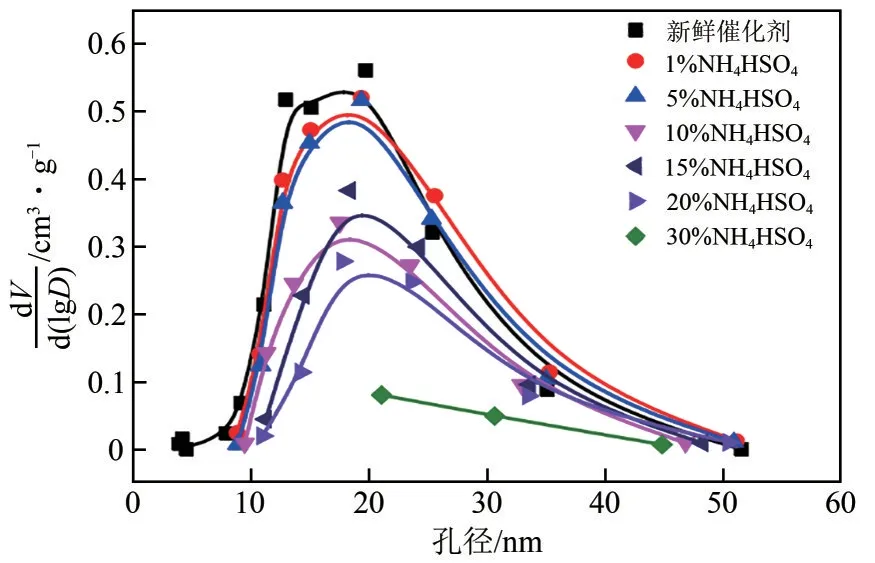
图1 负载不同质量 ABS 的催化剂孔径分布图[27]
3 钒钛系催化剂抗ABS中毒改进措施
为了提高钒钛系催化剂的抗ABS 中毒能力,需要采取有效的措施来减少催化剂表面ABS 的沉积量。现有研究一般从抑制催化剂表面ABS 产生、促进ABS 分解两方面入手,减少催化剂表面ABS的沉积,提高抗ABS中毒能力。
3.1 抑制催化剂表面ABS的生成
3.1.1 降低催化剂对SO2的氧化作用
ABS 的生成与SO3的浓度呈正相关[8],而SO2的氧化是SCR脱硝过程中SO3的主要来源之一,因此降低催化剂对SO2的氧化作用至关重要。在SCR脱硝过程中,有0.5%~1.5%的SO2被SCR 催化剂催化氧化成SO3[29]。研究发现,钒钛系催化剂中活性组分V2O5对SO2的催化氧化有明显的促进作用[30]。Kamata 等[31]对V2O5/TiO2催化剂上V2O5对SO2氧化活性的相关问题进行了研究。研究结果显示,添加的钒氧化物会均匀地分散在TiO2载体上,当V2O5负载量较低时,SO2的氧化速率与V2O5的负载量呈正相关;当负载量达到5.7%(质量分数)时,V2O5会完全覆盖TiO2载体形成一层膜;之后,随着负载量的进一步增加,SO2的氧化能力达到饱和。该学者进一步提出SO2的氧化行为可能会改变与V原子相连接的烃基V—OH,SO2与SO3的吸附与解吸过程中可能有V= = O与V—OH的参与。纪培栋[32]进一步研究发现,催化剂表面的SO2分别以SO2-3、HSO-3、SO2-4的形态吸附在V= = O、V—OH、Ti—OH 位点,V= = O 位点中的氧原子会转移至SO2中将吸附态的SO2氧化成为SO3,失去的氧原子在气相中的O2得到补充重新被氧化为V= = O。SO2在V2O5/TiO2催化剂上的氧化过程如图2所示。

图2 SO2在V2O5/TiO2催化剂上的氧化过程示意图[32]
Dunn 等[33]同样对V2O5/TiO2催化剂上V2O5对SO2氧化活性的影响进行了研究并提出了新的观点。他认为V2O5/TiO2催化剂中的Ti—O—V 化学键的氧原子能够吸附SO2,随后该氧原子转移到SO2中将其氧化成为SO3,原Ti—O—V化学键形成的氧空位由气相中的O2所补充,继而维持该催化氧化反应的不断进行。
(1)控制催化剂表面V2O5的负载量 V2O5/TiO2催化剂上SO2的氧化率与V2O5含量呈正相关线性关系,主要的原因是V2O5在催化剂表面的形态随着含量的增加从单体钒氧化物向多聚态的钒氧化物转变,甚至有V2O5晶体的形成[8,34],导致氧化位V5+= = O 的氧化性提高,SO2氧化能力提高。当V2O5在TiO2载体表面完全形成一层覆盖膜后,随着负载量进一步增加,SO2的氧化能力达到饱和[31,33]。SO2的氧化率与V2O5含量的关系见图3[35]。
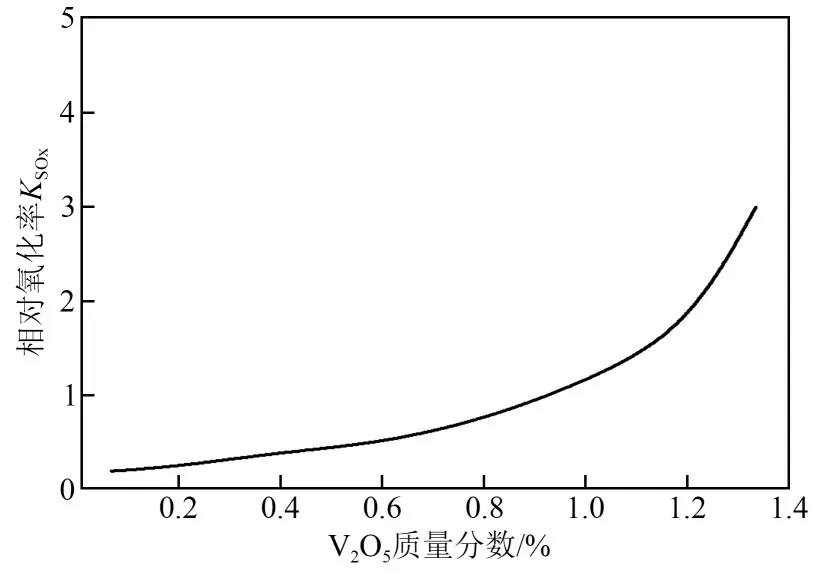
图3 SO2的氧化率与V2O5质量分数的关系[35]
在SCR脱硝过程中,当V2O5质量分数在0.8%~1.2%范围内时,V2O5/TiO2催化剂既有较高的脱硝效率,又有较低的SO2氧化率[22]。
(2)添加合适助剂 在钒钛系脱硝催化剂中添加不同的助剂能有效降低催化剂表面SO2的氧化性能。MoO3的掺杂能有效抑制V2O5/TiO2催化剂对SO2的氧化能力,当MoO3负载量大于9%时,催化剂表面SO2氧化率小于1%[36]。其主要作用机理如图4所示:V2O5/TiO2表面掺杂MoO3能有效抑制SO2和V= = O的反应,降低SO2的吸附,从而降低SO2的氧化率[37]。
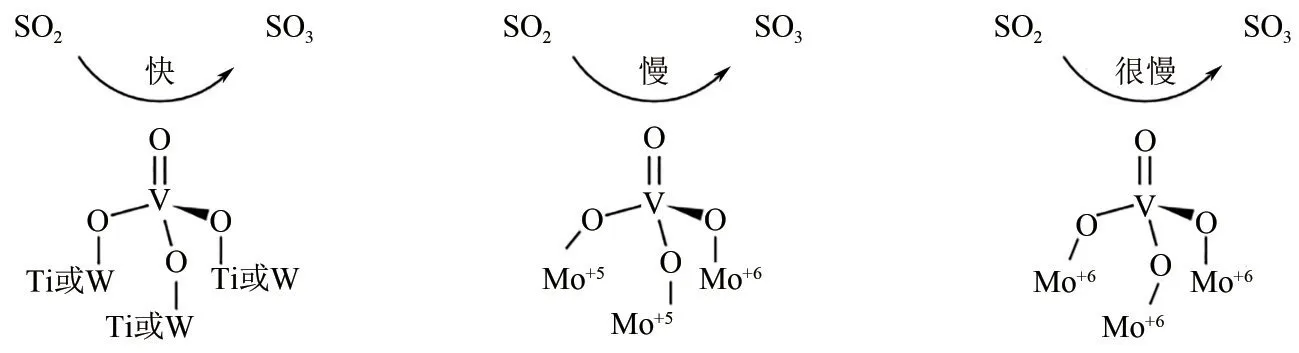
图4 V2O5−MoO3/TiO2降低SO2的氧化率作用机理[37]
此外,BaO 的添加会在催化剂表面形成一种V—O—Ba 结构,如图5 所示:该结构可抑制V2O5的氧化还原能力和SO2吸附能力,从而降低SO2的氧化效率,但脱硝活性也因此降低[38]。而对V2O5−BaO/TiO2催化剂进行硫酸化处理可在有效抑制SO2氧化的同时提升脱硝活性[38]。

图5 含W和BaO的V2O5催化剂在硫酸盐处理后的TiO2 (a)上和在无硫酸盐处理后的TiO2 (b)上[38]
掺杂Nb2O5后催化剂的比表面积小幅度降低将导致SO2吸附位点数量减少,从而SO2的吸附量下降,并且会降低V5+/(V5++V4+)比值和Oα含量,导致催化剂氧化还原能力下降,两者协同作用下Nb2O5的添加有助于大幅降低催化剂的SO2氧化率,当Nb 负载量为2%(质量分数)时,SO2氧化率最低[39−40]。Nb−V2O5−WO3/TiO2降低SO2氧化率的作用机理见图6。

图6 Nb−V2O5−WO3/TiO2降低SO2氧化率的作用机理[39]
(3)优化催化剂物理结构 合理调控催化剂壁厚能有效降低催化剂表面SO2的氧化。SCR 反应外扩散发生在催化剂表面壁厚的75~100μm 内;而SO2氧化为SO3属于化学反应,在整个催化剂壁厚内进行[5,41−43],见图7。分布在壁面中间区域的活性组分不参与脱硝反应但能促进SO2的氧化。Schwämmle 等[44]研究发现蜂窝式结构催化剂的壁厚与SO2氧化率呈正相关线性关系,后续赵大周等[45]利用Fluent软件模拟也证实了这个观点,当催化剂壁厚从0.6mm 增加到1.2mm 时,催化剂表面的SO2氧化率从0.5%增加到1%。张静等[46]将蜂窝式脱硝催化剂的壁厚控制在0.66mm,发现其表面的SO2氧化率较低。
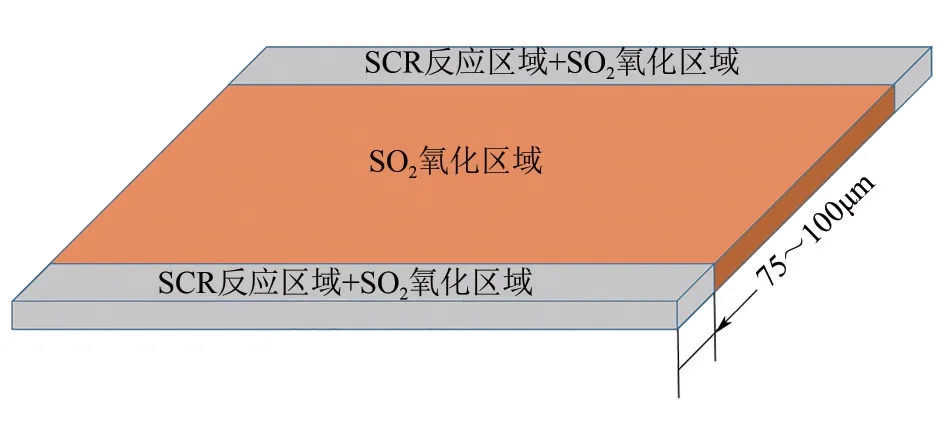
图7 催化剂不同壁厚反应类型
此外,合理地调控催化剂的孔结构,降低SO2在催化剂内部的渗透也是降低催化剂表面SO2氧化率的有效措施[42]。唐坚等[47]制备了一种平均孔径仅为15~20nm、表面微孔深度低于200μm 的SCR 脱硝催化剂,活性测试结果显示该催化剂脱硝效率略高于市售催化剂,但SO2氧化率仅为市售催化剂的一半左右。催化剂表面微孔孔径的减小以及孔深的缩短能在微观上减小SO2氧化区域的范围,促进NH3向催化剂微孔中的扩散,提高脱硝反应在微孔内所占的比例,从而降低SO2的氧化率。
在催化剂表面构建一层多孔结构的隔离层同样能抑制催化剂表面SO2的氧化。贾媛媛等[48]以正硅酸乙酯为前体在催化剂表面附着了一层多孔无机硅,测试结果显示SO2氧化率由85%降到了41%。多孔结构的隔离层能有效抑制反应组分与催化剂活性位点之间的接触,从而抑制副反应的发生;烟气中SO2与催化剂活性中心的接触行为被抑制,SO2氧化率因此降低。
3.1.2 抑制VOSO4物种的产生
SO2与钒钛系催化剂表面的V2O5反应后生成金属硫酸盐(VOSO4),NH3会与VOSO4反应而生成ABS,通过对钒钛系催化剂进行改性来抑制催化剂表面金属硫酸盐(VOSO4)的生成是抑制ABS在催化剂表面生成的有效措施。研究发现,在催化剂表面掺杂金属氧化物能有效抑制催化剂表面金属硫酸盐的产生。其中Fe2O3因其优异的织构性能和极具竞争力的价格在催化剂领域被广泛应用。如图8所示,Fe2O3与V2O5−WO3/TiO2机械混合的催化剂中,SO2可以直接吸附到Fe2O3上,从而减少其在氧化钒上的吸附;且钒位点上氧化生成的SO3也可被吸附在Fe2O3上而形成FeSO4,从而通过减少VOSO4来抑制ABS的生成[49]。V2O5−WO3/TiO2+Fe2O3催化剂具有较高的比表面积和孔体积,且FeSO4的形成可提供额外的Brønsted酸位点,可使其在低温下具有最佳的NH3−SCR活性和抗硫性能。
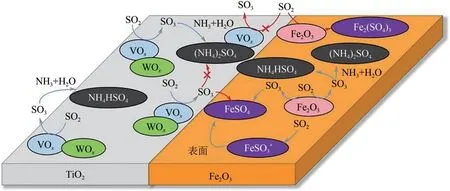
图8 V2O5−WO3/TiO2+Fe2O3催化剂表面硫酸盐生成机理[49]
CeO2掺杂到V2O5−WO3/TiO2催化剂中后,其对活性V位点同样具有保护作用,可提高NOx吸附的稳定性并有效抑制SO2的吸附,从而阻止VOSO4的形成[50−51]。此外,V 的电子通过氧化还原传递给CeO2,使CeO2以富电子态存在,促使CeO2周围的电子向硫酸盐迁移,增强CeO2与ABS之间的结合,从而削弱ABS 的稳定性,促进ABS 中NH4+与NO、O2之间的NH3−SCR反应。对于V2O5−CeO2/TiO2催化剂,有研究发现其表面的ABS 在加热分解过程中会生成更难分解的Ce2(SO4)3,如图9 所示,从而抑制催化剂表面ABS 的分解行为[26]。对此,有学者[52]在V2O5−CeO2/TiO2催化剂中掺杂WO3,研究发现催化剂表面形成了一种W—O—Ce结构,该结构可加强W、V 和Ce 物种之间的相互作用,提高它们的氧化还原能力,在催化剂上形成双重氧化还原循环,如图10 所示。双重氧化还原循环的形成降低了催化剂对SO2的吸附作用,抑制了VOSO4物种的形成;并通过电子转移的协同作用从相邻的V和W捕获电子来增加Ce3+数量,使Ce 物种与ABS 之间的电子向Ce 方向发生偏移,从而削弱ABS 在催化剂上的稳定性,使NO+O2与ABS 发生NH3−SCR 反应。在5%(质量分数)WO3负载下,脱硝转化率较快,ABS的形成被完全抑制。因此,WO3的添加对催化剂表面ABS的分解同样具有重要作用。
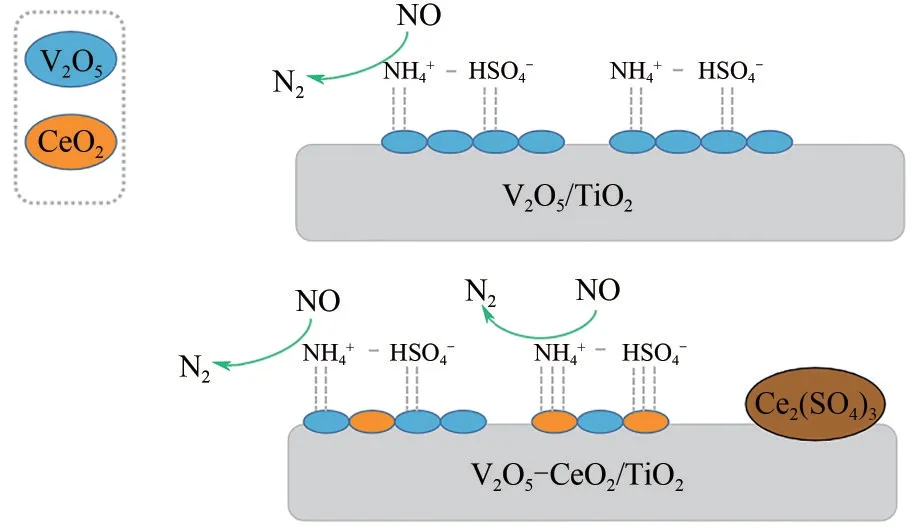
图9 CeO2掺杂对ABS分解和反应行为的影响[53]
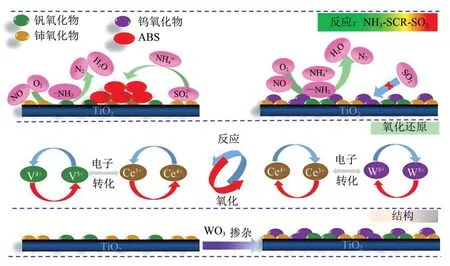
图10 WO3质量分数为5%时抗ABS性能较高的构效关系[52]
3.2 促进催化剂表面ABS的分解
在SCR 脱硝过程中,提高催化剂表面ABS 分解能力能有效提高催化剂抗ABS中毒能力。ABS在钒钛系催化剂表面沉积时与TiO2相互作用形成NH4+和双齿SO24-,其中NH+4稳定性较低,沉积在催化剂表面;双齿SO24-中Ti原子和S原子中的电子转移到O原子中,抑制了S元素的还原,延缓了ABS分解为SO2的过程[54]。与纯ABS物质相比,沉积在催化剂表面的ABS具有更高的热稳定性,分解难度大大增加。钒钛系催化剂表面ABS分解通常分为两步:第一步为脱氨过程,沉积在催化剂表面的NH+4率先分解,分解温度在250℃左右;第二步为硫酸盐物种分解过程,催化剂表面双齿SO2-4发生分解释放SO2,分解温度在350℃左右[54]。因此,加速消耗NH+4、高效还原SO2-4能有效促进催化剂表面ABS的分解。
(1)加速NH+4的消耗 现有研究显示,在SCR反应过程中催化剂表面ABS与NO两者之间存在反应[55],该反应减少了催化剂表面ABS的沉积量,能在一定程度上避免SCR 反应过程中催化剂ABS 中毒问题。其反应机理与SCR 机理类似:ABS 中的NH+4作为还原剂与NO在活性位点V= = O上发生氧化还原反应,产物为N2和H2O;V2O5在NH+4与NO 的反应中起关键作用,且V2O5/TiO2催化剂表面V2O5负载量越多,越有利于该反应的进行[56]。具体反应路径见图11。
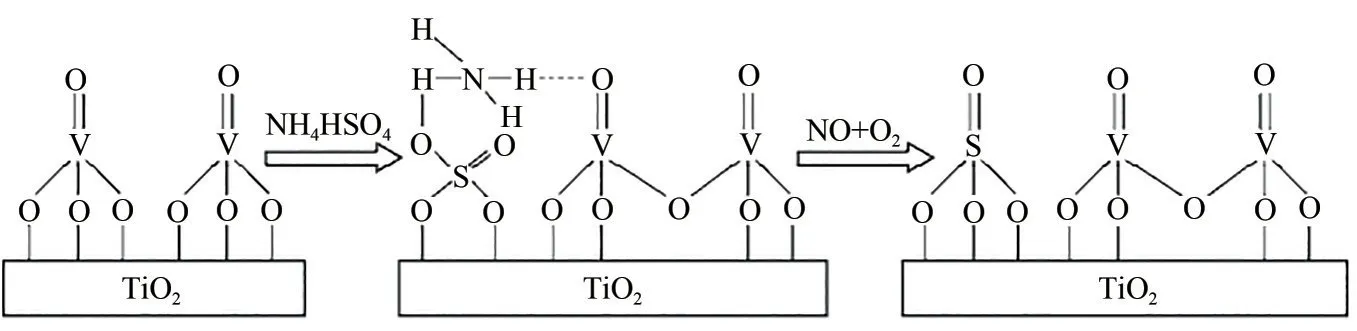
图11 V2O5/TiO2催化剂作用下NO与ABS的反应路径[56]
SiO2是一种比表面积较大、化学性质相对稳定的物质,具有较高的熔点和沸点。将SiO2掺杂到TiO2中能增加SCR催化剂表面活性位点酸性、改善催化剂物理结构并提高催化剂热稳定性[57−58]。ABS在催化剂表面沉积后,与TiO2表面的NH4+相比,TiO2−SiO2复合载体上的NH+4 在加热过程中更容易被消耗,从而导致催化剂表面ABS 快速分解。分解路线如图12所示[59]。

图12 ABS在TiO2和TiO2−SiO2载体上的分解路线[59]
对于复合载体的V2O5/TiO2−SiO2−MoO3催化剂而言,其具有更多的固体酸性位点,能进一步促进ABS 中NH+4与NO 的反应,加速了NH+4的消耗,从而促进催化剂表面ABS 的分解[60]。在SO2存在的条件下,V2O5−MoO3/TiO2−CeO2催化剂掺杂SiO2能促进表面ABS的热分解,提高低温下V2O5−MoO3/TiO2−CeO2与NO的反应活性[61]。
活性组分Mo 在V2O5/TiO2催化剂中的负载可使ABS 解离后的NH+4吸附在Lewis 酸位点上,促进ABS中NH+4与NO的反应,提高ABS分解速率[26,62]。
(2)促进SO2-4的还原 研究发现,V2O5能促进催化剂表面含硫化合物的还原,适当调节钒钛系催化剂表面V 含量有利于抑制ABS 在TiO2上的吸附,加速含硫化合物的还原,从而降低催化剂表面ABS 的分解难度[12,62]。当V2O5/TiO2催化剂表面V 含量较少时,ABS会优先吸附在催化剂载体TiO2上并产生NH+4和热稳定性较高的双齿状SO2-4;当V含量[4.5%(质量分数)]足以覆盖催化剂载体TiO2时,V2O5的存在可以在钒钛系催化剂表面形成一层保护层,ABS中的SO2-4不再与TiO2位点相结合,而是形成类似VOSO4的结构,该结构与双齿状SO2-4相比热稳定性较低,导致ABS 更易分解,分解过程与纯ABS相似[54]。分解过程示意图见图13。
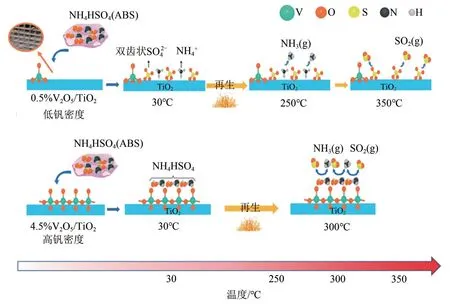
图13 ABS在0.5%V/Ti和4.5%V/Ti催化剂上的分解过程示意图[54]
WO3的掺杂可以增大SO2-4中S原子周围的电子云密度,促进ABS中+6价的S原子还原为SO2中+4价的S 原子,从而促进ABS 的分解[63]。V2O5/TiO2和V2O5−WO3/TiO2催化剂上ABS 的分解过程中电子转移情况如图14所示。进一步添加Sb2O5和Nb2O5后,可增加硫酸盐中S 原子周围的电子云密度,促进ABS和NO的反应,增强低温时SO2-4向SO2的转变,使ABS在催化剂上的分解行为明显增强[64]。
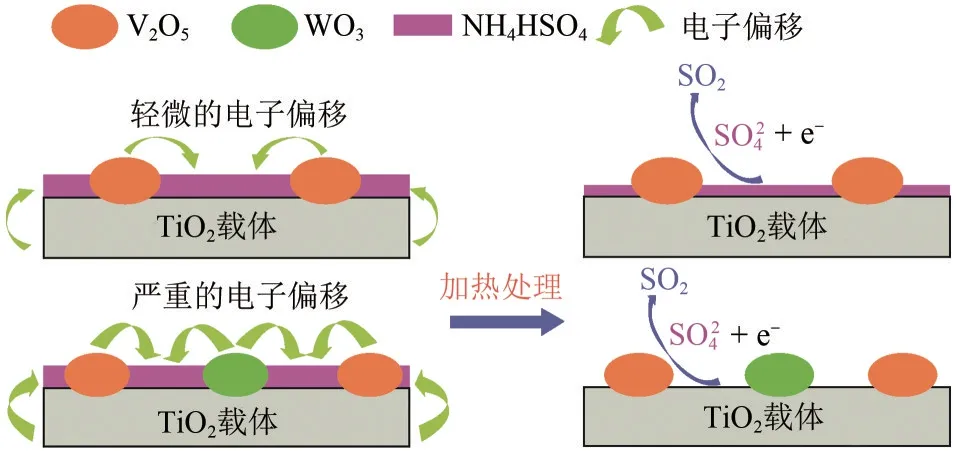
图14 V2O5/TiO2和V2O5−WO3/TiO2催化剂上ABS的分解[30,65]
4 结语
本文介绍了ABS 在SCR 脱硝过程中的生成机理,探讨了ABS 在催化剂表面的沉积机理、影响因素及危害,并对现阶段钒钛系催化剂抗ABS 中毒改性研究作了总结,主要结论如下。
(1)烟气中ABS 生成路径主要有两种,即气相反应机理和催化剂表面反应机理。当温度低于300℃时,ABS 主要按气相两步反应机理进行。受反应温度、NOx浓度、NH3/NO 体积比、NH3/SO3摩尔比等因素的影响,ABS在催化剂内的沉积量与性状均有所不同,主要沉积于催化剂孔道结构中。
(2)钒钛系催化剂抗ABS 中毒改进措施主要集中于抑制催化剂表面ABS 产生和促进ABS 分解两方面。调整催化剂表面V2O5含量、优化催化剂物理结构、添加合适的助剂都是提高钒钛系催化剂抗ABS中毒的有效措施。
(3)研究表明,NO 浓度对催化剂表面ABS 有双重作用,NO 既能加速钒钛系催化剂表面SO2的氧化,加速ABS 生成,又能和催化剂表面ABS 发生反应,加速铵根离子的消耗而促进ABS 分解。但实际烟气中NO浓度常由燃料特性与运行工况控制,较难调控。现有钒钛系催化剂抗ABS 改性常关注化学特性调控,主要集中于Ce、Mo 等活性组分负载,后续研究应综合调控催化剂物理结构与化学结构,通过控制壁厚、孔径以及加装隔离层等物理结构优化方式来抑制ABS 沉积,并通过相关助剂的添加抑制SO3生成、促进沉积后ABS分解,最终有效实现催化剂抗ABS 中毒性能,提高催化剂低温脱硝活性。
