二氧化碳与环氧化合物催化共聚反应研究进展
2023-02-22姜奕彤陈欣月吕洪兵王子春
*姜奕彤 陈欣月 吕洪兵 王子春
(北京化工大学 北京 100029)
通过CO2捕集、封存与利用技术实现CO2资源化利用的途径主要有:(1)利用CO2的物理性质,如CO2驱油技术等。(2)通过生物或化学转化的方法,将CO2转化为甲醇、燃料和聚碳酸酯材料等高附加的化学品。高活性环氧化合物能够在较温和条件下与CO2共聚合成CO2基生物可降解聚碳酸酯(PC)。PC是能够在自然环境中完全生物降解的塑料,可用于一次性包装材料、餐具、保鲜膜、一次性医用材料、地膜等方面。PC中的CO2含量可达31%~50%,不但可以大幅降低聚酯合成中对上游石化原料的需求、降低碳排放,而且能够为解决石油基废旧塑料造成的“白色污染”提供替代方案,降低环境中微塑料颗粒对人类和野生动物健康的危害。开发高活性、高选择性催化体系是实现PC高效合成的关键。本文主要对PC开发中高性能均相催化体系的研究进展进行了系统性评述。
1.基础催化机理
1969年,Inoue等人[1]发现ZnEt2能够催化CO2与环氧乙烷共聚合成聚碳酸酯。此后,多种催化体系相继被开发出来。共聚反应通过配位-插入机理进行(如图1所示)。
首先是路易斯酸活化环氧化合物,活化的环氧化合物被亲核试剂攻击开环并与之形成金属醇盐;随后CO2插入该中间体形成金属碳酸盐。在不断增长的阴离子链中交替引入环氧化合物和CO2实现链增长,得到交替排列的PC。但实际PC合成过程通常会发生分子链末端金属烷氧基进攻链上重复单元,生成五元环碳酸酯单体,这是由分子内链回咬造成的环加成反应;也会发生环氧化物均聚反应,在聚碳酸酯共聚链上出现聚醚重复单元;外部质子源的存在终止当前聚合,发生链转移,形成羟基封端共聚物,新的金属烷氧基活性中心继续聚合,分子链数目增加,共聚物分子量降低。因此,通过设计合理催化体系、调控活性中心间竞争协同机制,开发高活性、高产物选择性、高稳定性的催化体系是实现CO2与环氧化物共聚反应高效合成PC以及控制PC结构和性能的关键。
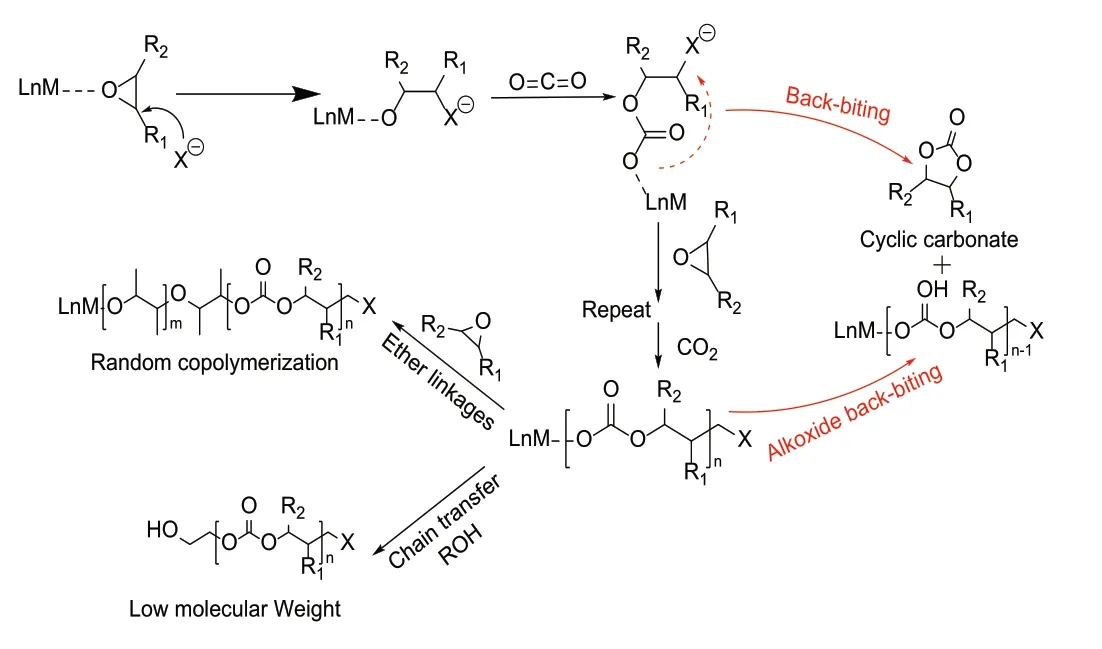
图1 CO2和环氧化合物共聚可能涉及的反应机理
2.催化体系
(1)金属催化剂
①金属卟啉配合物催化体系
Taketa和Inoue[2]首次合成了四苯基卟啉铝配合物用于催化环氧丙烷和CO2交替共聚,但耗时长。Cooper等人[3]开发的铬的氟化卟啉配合物在DMAP的协同作用下得到含量为90%~97%碳酸酯键的聚合物,每克铬能够产生3.9kg的聚合物)。近期,Miller等人[4]系统性研究了金属卟啉配合物上不同的金属中心以及不同的配体取代基团的影响,发现带有吸电子取代基的钴中心卟啉配合物催化性能最好,其转化频率(TOF)是铝作为金属中心的两倍。
②金属-β-二亚胺催化体系
Coates等[5]开发了单/双核β-二亚胺化锌催化剂并合成了含碳酸酯键95%的PC。Allen[6]研究取代基和引发基团对催化活性的影响,发现大位阻取代基的配合物表现出更高催化活性;主链上引入吸电子基团会进一步提高活性。但锌配合物催化体系难以抑制环状碳酸酯的生成,催化PO与CO2的活性较低,且选择性只有75%。在双核催化体系研究中发现第一种金属参与环氧化合物预配位,第二种金属加入生长的共聚物链,从而使环氧化合物开环,具有更高的活性[7]。
③双金属配合物
双金属催化体系能够为不断增长的聚合物链和环氧化合物单体提供相邻的结合位点,从而抑制单核金属催化体系中“自由”生长的聚合物链的分子内回咬问题,减少环状碳酸酯的生成。Lee等人[8]设计了苯胺基锌配合物,发现吸电子取代基能够降低金属中心的电子密度,有助于CO2/环氧化合物的聚合,并制得摩尔质量高达8万~28万g·mol-1的聚环己烯碳酸酯(PCHC)。Williams等人[9]设计“还原型Robson”大环配体的双金属锌配合物高效合成PCHC,并发现大环状配体和双金属结构是高活性的关键。Xiao等人[10]合成Trost酚盐配体的同核双金属配合物催化环氧环己烯(CHO)/CO2共聚,发现同等条件下双镁是双锌催化剂活性的两倍。Kember等人[11]用Co来取代Zn形成双金属配合物,发现由于碳酸钴链增长的亲核性增加,能够加快速控步骤反应速度,从而提高了其催化CHO/CO2共聚的活性。
与上述同核双金属体系相比,异核双金属中两种金属中心存在不同作用的“链式穿梭机制”,具有更高的共聚反应催化活性。Williams等人[12]设计了Mg/Zn配合物,其活性是双Mg催化剂的两倍,并揭示了金属间协同作用增强共聚催化活性的机制。在Zn与第1,2,13族金属形成的异核双金属配合物中,该团队发现除Mg/Zn外,大多数异核金属间缺乏协同作用、活性低[13]。该团队又开发了镁钴双金属催化剂,由于镁与CHO配位的能垒增强了过渡态熵值,钴增强中间体碳酸钴亲核性从而降低了过渡态焓值,其活性远高于双Co以及双Mg催化剂,是Zn/Mg异核双核催化剂活性的4倍[14]。
④席夫碱类催化体系
席夫碱类(Salen)催化体系是催化CO2以及环氧化合物活性最高的催化体系之一。研究最广泛的是以Cr、Co为金属中心的催化体系。Jacobsen等人[15]发现手性铬席夫碱配合物是生成聚碳酸酯的有效催化剂。受Jacobsen启发,Darensbourg团队设计出活性、立体选择性和稳定性都更高的席夫碱铬催化体系[16]。还原(Salan)及半还原(Salalen)的席夫碱配体衍生物也被用作催化剂配体替代Salen来改善催化剂性能。Rao等人[17]设计了[SalanCrX]催化剂在DMAP协同作用下,其催化活性是相同Salen体系的30倍。与Salan和Salen不同,Salalen催化体系中配体并不保持在刚性平面,可以在轴向和赤道位置结合,这种配位柔性促进生长中的碳酸酯链双齿结合,降低CO2插入能垒并抑制脱羧,从而产生具有99%碳酸酯键的共聚物。Nozaki等人[18]研究了异核双金属席夫碱催化体系,高负载下TOF为70h-1,生成含有99%碳酸酯键的PPC,选择性>99%。与单金属配合物相比,双金属催化活性显著提高,但区域选择性略低。该研究团队还发现将配体修饰为内部含有助催化剂的结构,能够显著增强催化共聚的选择性,同时也利于催化剂的回收利用[19]。
(2)非金属催化剂
聚碳酸酯产品中潜在有毒微量金属的问题限制了其在生物医学中的应用,因此,有机催化CO2与环氧化物共聚合成生物基可降解PC受到了越来越多的关注。有机催化体系最为常见的是由有机硼和鎓盐或有机碱协同使用的路易斯酸碱对。Boopathi等人[20]合成出三乙基硼烷和各种鎓盐组成的有机催化体系,用于PO或CHO与CO2的交替共聚。Wu等人[21]通过将硼烷铵盐以比例整合进入一个分子内构建动态路易斯多核系统(DLMCS),设计了一系列单核、双核、三核和四核有机硼催化剂,并发现硼与铵盐之间的分子内协同效应,在介导催化活性和选择性方面起着关键作用;而且这些催化剂可以通过精密调控路易斯酸性硼中心的亲电性质、正电铵阳离子的电子和空间效应、硼中心和铵阳离子之间的连接体长度、硼中心的数量和亲核阴离子来调控催化活性与选择性。该团队开发的风车状四核有机硼催化体系,在温和条件下催化环氧氯丙烷和CO2共聚具有>99%的PPC选择性。但与金属催化剂相比,有机催化剂催化的活性和选择性仍偏低。
3.总结与展望
CO2与环氧化物的共聚是制备新型可持续聚合物材料实现CO2大规模资源化利用的一条极具潜力的技术路线,具有解决塑料“白色污染”和降低二氧化碳排放的巨大潜力。研究表明,基于席夫碱催化体系和β-二亚胺催化体系的双金属配合物,具有比单金属配合物更高的活性和选择性。基于此,探索以廉价且储量丰富的金属开发双金属配合物催化剂具有重要的研究价值和应用潜力。此外,配体的结构、与助催化剂的协同作用都能够显著影响金属的催化活性。因此,通过理论计算手段合理设计配体结构、开发改性方法实现席夫碱与内部助催化剂协同作用的精准调控,能够为高性能共聚催化剂的开发提供重要的理论基础和技术方法。在此基础上,开发环境友好型的无金属有机催化体系,实现绿色、无害化的聚碳酸酯合成技术路线的开发,对于满足医用材料等特殊需求和可持续发展具有重要的意义。
