HPLC法测定注射用头孢呋辛钠有关物质方法的优化
2023-02-22陈仲祥林美龄尹可欣熊莉平
*陈仲祥 林美龄 尹可欣 熊莉平
(1.重庆工信职业学院 重庆 401233 2.西南药业股份有限公司 重庆 400038)
注射用头孢呋辛钠为《中国药典》[1]收载品种,根据2020年5月14日国家药监局发布的关于开展化学药品注射剂仿制药质量和疗效一致性评价工作的公告(2020年第62号)[2],对其进行一致性评价。注射用头孢呋辛钠为原料药无菌分装制得,根据本品原料合成工艺路线可能引入杂质,对各国药典(ChP2020、JP18、BP2020、EP10.0、进口注册标准JX20170207)中提及杂质及本品稳定性试验产生杂质进行分析,对本品杂质谱进行梳理,汇总本品可能存在杂质为:杂质A、杂质B、杂质C、杂质E、杂质F、杂质G、杂质H、杂质I、杂质8、杂质9及杂质11,对其进行研究。将上述可能产生或存在的杂质配制成系统适用性溶液筛选合适的方法,结果各国药典方法均未能同时将上述杂质基线分离或杂质洗脱不完全。故在中国药典方法上进行优化,延长了最终比例流动相的洗脱时间,更好地保证流动相的洗脱能力,取得理想的试验结果,现报道如下。
1.材料
(1)主要仪器
XPR2电子天平(梅特勒);ME204T/02电子天平(梅特勒);XS105DU电子天平(梅特勒);BT25S电子天平(赛多利斯);KH-500DB数控超声波清洗器(昆山禾创);PB-10酸度计(赛多利斯);LC-20AT高效液相色谱仪(岛津)。
(2)试药
注射用头孢呋辛钠(西南药业股份有限公司,批号:Y200601,规格:1.5g);西力欣(GSK,批号:PL7S,规格:0.75g);头孢呋辛对照品(中检院,批号:130493-201706,纯度:91.9%);杂质A对照品(TLC,批号:2413-038A3,纯度:95.9%);杂质B对照品(TLC,批号:2469-088A5,纯度:93.7%);杂质C对照品(TLC,批号:2469-066A9,纯度:97.4%);杂质E对照品(QCC,批号:2469-066A9,纯度:96.24%);杂质F对照品(QCC,批号:18-SEP-18-12,纯度:95.53%);杂质G对照品(TLC,批号:3422-069A11,纯度:95.2%);杂质H对照品(TLC,批号:2277-014A4,纯度:97.9%);杂质I对照品(TLC,批号:2756-091A4,纯度:96.8%);杂质8对照品(TLC,批号:3278-001A2,纯度:99.0%);杂质9对照品(TLC,批号:3218-096A4,纯度:97.5%);杂质11对照品(TLC,批号:2405-090A2,纯度:97.8%);甲醇、乙腈为色谱纯;冰醋酸、冰乙酸、三水合乙酸钠等其余试剂均为分析纯;水为超纯水。
2.方法与结果
(1)色谱条件
色谱条件用辛烷基硅烷键合硅胶柱为填充剂(推荐Agilent Zorbax SB-C8柱,4.6mm×150mm,5μm或效能相当的色谱柱);流动相A为醋酸盐缓冲液(取醋酸钠0.68g,冰醋酸5.8g,加水稀释至1000mL,用冰醋酸调节pH值至3.4);流动相B为乙腈;流速为每分钟1.5mL,按表1进行线性梯度洗脱;检测波长为273nm;柱温为25℃;进样体积20μL。
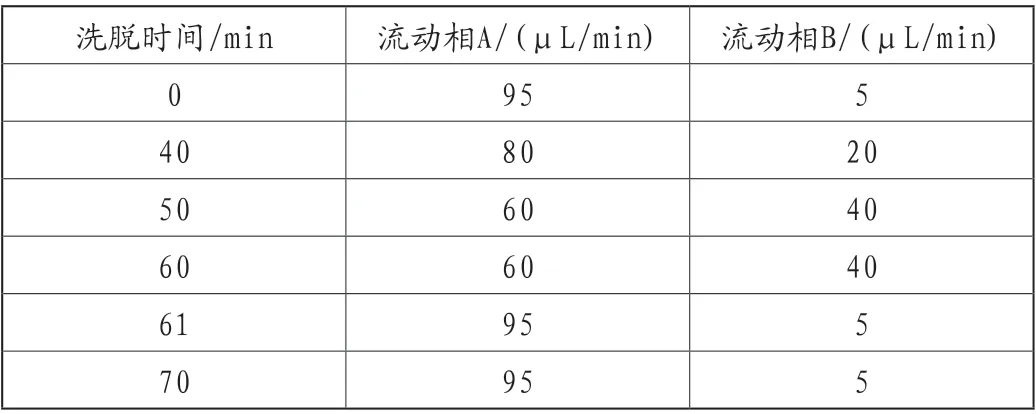
表1 有关物质梯度洗脱
(2)溶液的配制
溶剂:水。供试品溶液:含头孢呋辛钠约0.5mg/mL的溶液。
对照溶液:精密量取供试品溶液适量,用水定量稀释制成每lmL中含1μg的溶液。
杂质储备液:精密称取杂质A、杂质B、杂质C、杂质E、杂质F、杂质G、杂质H、杂质I、杂质8、杂质9、杂质11对照品各适量,分别置10mL量瓶,加90%乙腈超声溶解并稀释至刻度,摇匀。
杂质定位溶液:分别精密量取上述杂质储备液1mL,分别置不同的20mL量瓶中,加水稀释至刻度,摇匀。
杂质与样品混合溶液:精密称取头孢呋辛对照品约5mg置50mL量瓶中,精密加入上述杂质母液:杂质A(0.8mL)、杂质B(0.8mL)、杂质C(0.4mL)、杂质E(0.8mL)、杂质F(0.9mL)、杂质G(0.8mL)、杂质H(0.8mL)、杂质I(0.2mL)、杂质8(0.4mL)、杂质9(1.0mL)、杂质11(0.2mL),加水溶解并稀释至刻度,摇匀,作为杂质与样品混合溶液。
(3)系统适用性试验
精密量取稀释剂、上述杂质定位溶液、杂质与样品混合溶液各20μL,分别注入液相色谱仪,记录色谱图。见图1,结果显示各杂质能有效分离,空白溶剂不干扰测定,专属性符合要求。
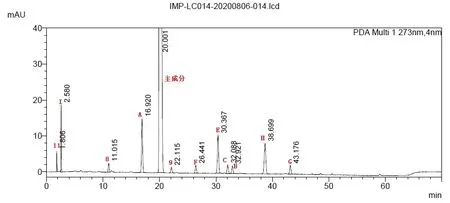
图1 方法专属性色谱图
(4)检测限和定量限
用溶剂逐步稀释各已知杂质对照品储备溶液,获得的色谱图中,信噪比约为10∶1时,确定为定量限;信噪比约为3∶1时,确定为检测限。各杂质定量限远低于控制限度要求,表明本方法能满足杂质检测灵敏度的要求,具体详见表2。
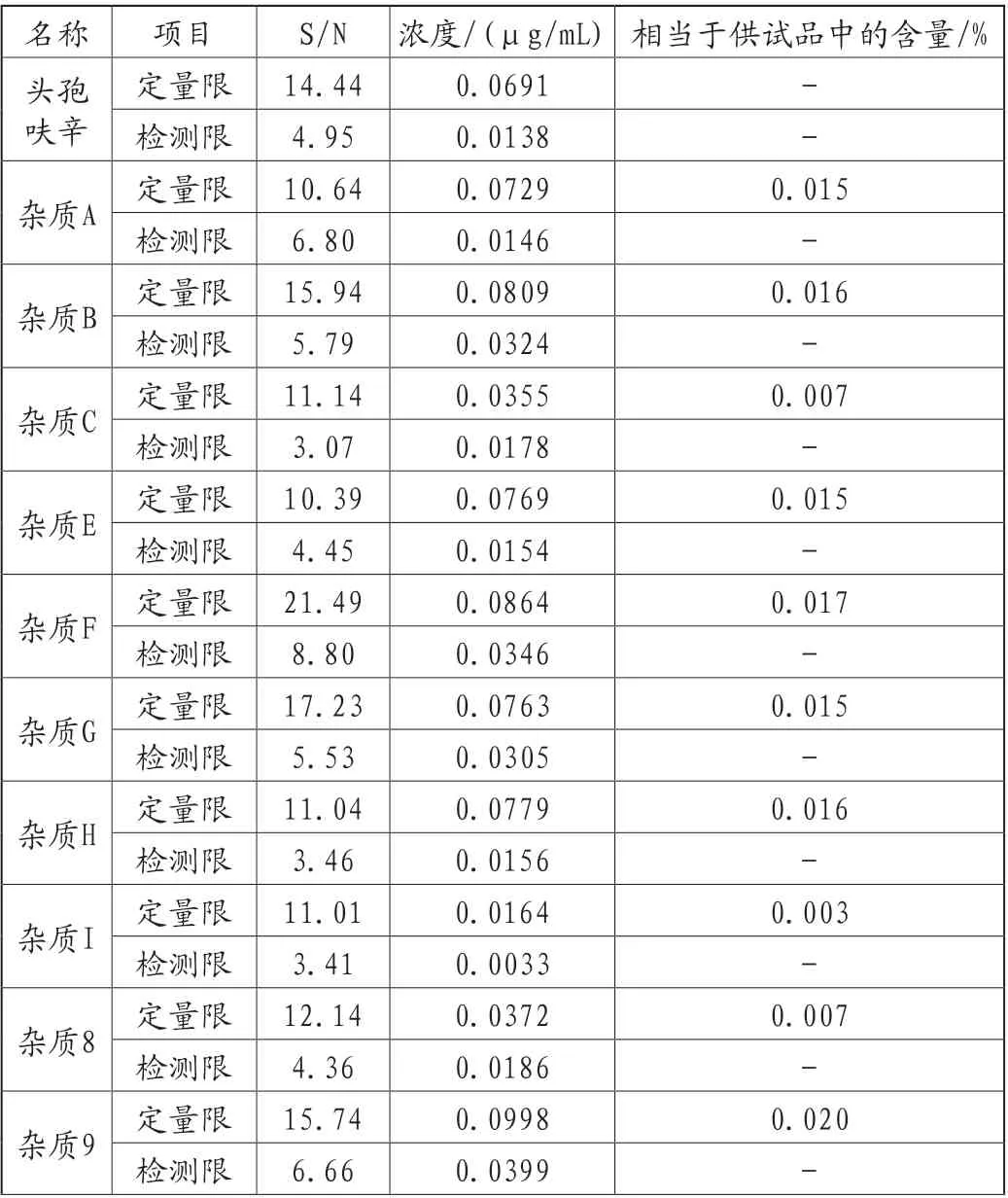
表2 定量限和检测限结果

续表
(5)线性和校正因子
在定量限到150%限度水平配制线性溶液进样,以峰面积为纵坐标(y)、进样质量浓度为横坐标(x)进行线性回归,并按以下公式计算校正因子,校正因子=头孢呋辛线性方程的斜率/杂质线性方程的斜率。线性范围和校正因子考察结果详见表3。

表3 线性范围和校正因子考察结果
(6)精密度
由试验员A在A时间采用A仪器,平行测定6份供试品,作为重复性结果。由试验员B在B时间采用B仪器平行测定6份供试品,合并重复性样品共12份样品作为精密度结果。12份RSD值评价不超过20%,精密度良好,结果详见表4。
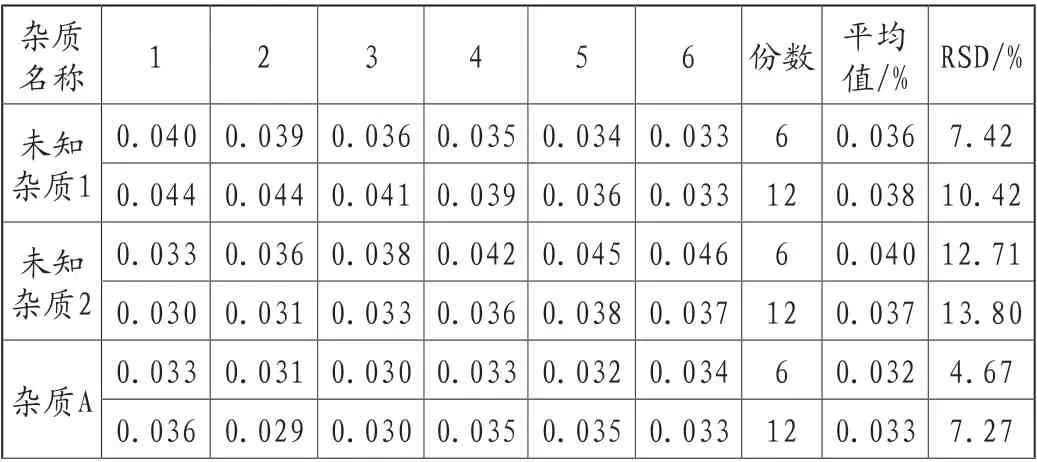
表4 精密度考察结果
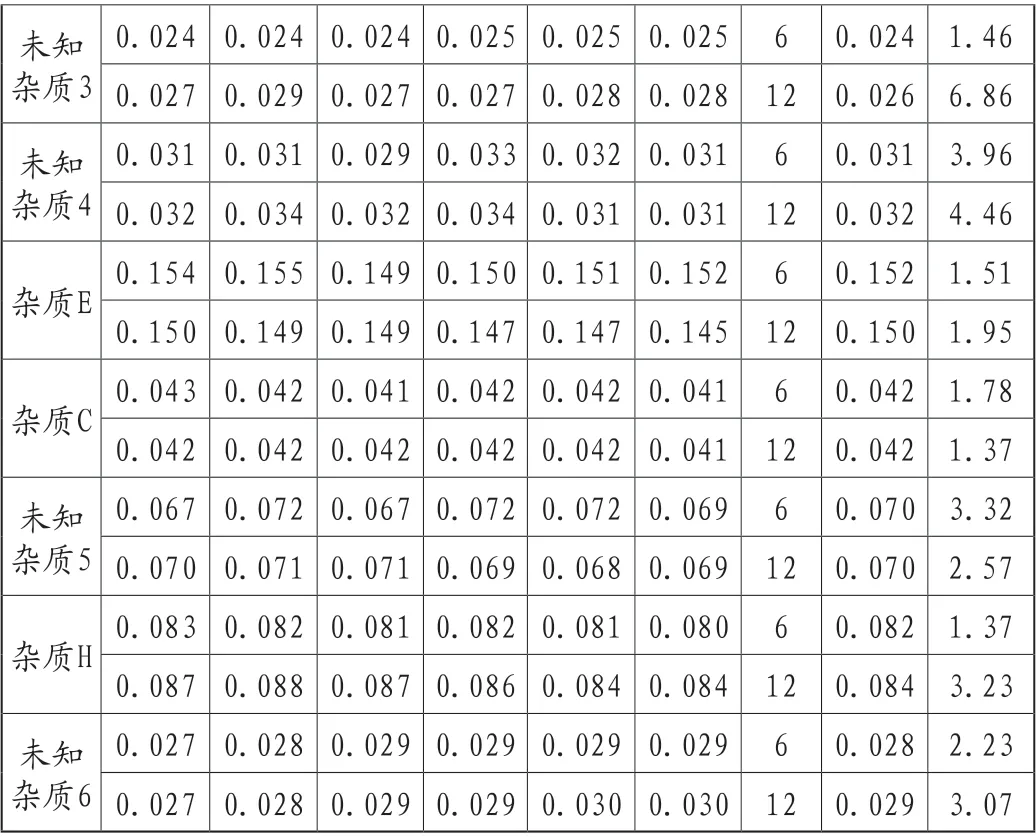
未知杂质3 0.024 0.024 0.024 0.025 0.025 0.025 6 0.024 1.46 0.027 0.029 0.027 0.027 0.028 0.028 12 0.026 6.86未知杂质4 0.031 0.031 0.029 0.033 0.032 0.031 6 0.031 3.96 0.032 0.034 0.032 0.034 0.031 0.031 12 0.032 4.46杂质E 0.154 0.155 0.149 0.150 0.151 0.152 6 0.152 1.51 0.150 0.149 0.149 0.147 0.147 0.145 12 0.150 1.95杂质C 0.043 0.042 0.041 0.042 0.042 0.041 6 0.042 1.78 0.042 0.042 0.042 0.042 0.042 0.041 12 0.042 1.37未知杂质5 0.067 0.072 0.067 0.072 0.072 0.069 6 0.070 3.32 0.070 0.071 0.071 0.069 0.068 0.069 12 0.070 2.57杂质H 0.083 0.082 0.081 0.082 0.081 0.080 6 0.082 1.37 0.087 0.088 0.087 0.086 0.084 0.084 12 0.084 3.23未知杂质6 0.027 0.028 0.029 0.029 0.029 0.029 6 0.028 2.23 0.027 0.028 0.029 0.029 0.030 0.030 12 0.029 3.07
(7)加样回收率
精密称取注射用头孢呋辛钠(Y200601批,1.5g)内容物约12.5mg,置25mL量瓶,称取9份,分别加入杂质限度50%、100%、150%的杂质对照溶液,加水稀释至刻度,摇匀(临用新制);分别精密量取20μL,注入液相色谱仪。记录色谱图,平均加样回收率在98.75%~109.1%范围内,RSD在1.29%~4.81%范围内,表明本方法准确度良好,详见表5。
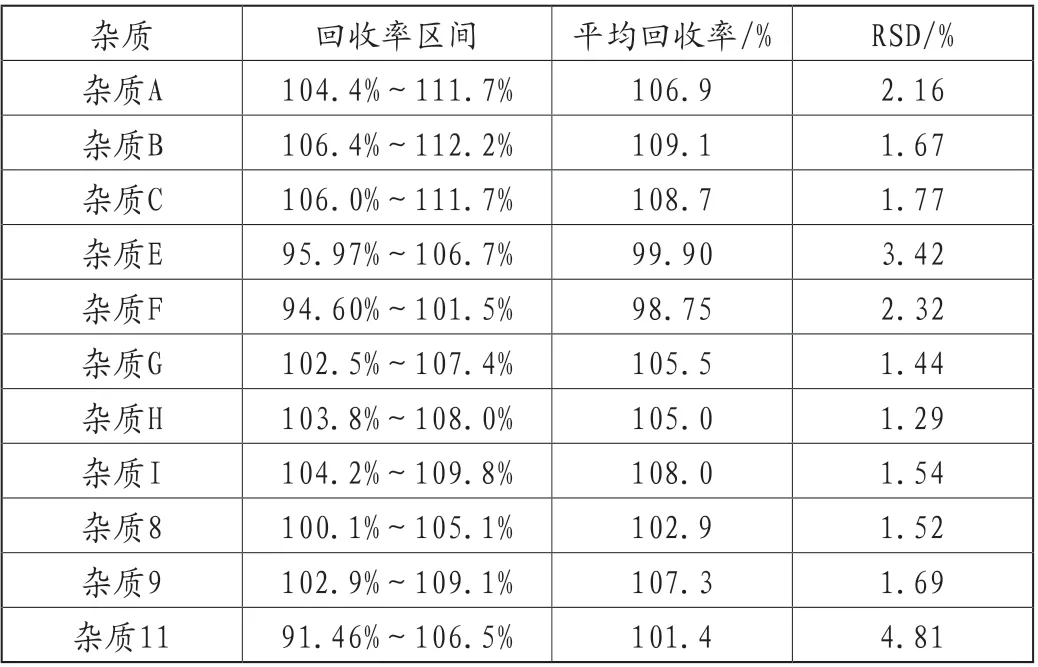
表5 回收率考察结果
(8)耐用性
耐用性实验中对流速(±0.1mL/min)、柱温(±2℃)、色谱柱批号、初始流动相比例(±2)、pH(±0.05)、波长(±2nm),微调色谱条件后,主峰与相邻杂质峰分离度大于1.5,各相邻杂质间均可有效分离;柱温在23~25℃,样品溶液中杂质检出个数及检出量基本一致,即建议柱温控制在此温度范围内;流速在1.4~1.5mL/min时,样品溶液中杂质检出个数及检出量基本一致,即建议流速控制在此范围内;pH在3.35~3.40范围内,样品溶液中杂质检出个数及检出量基本一致,即建议流动相pH控制在此范围内;有机相比例变动±2%、波长变动±2nm及更换不同批次色谱柱时,样品溶液中杂质检出个数及检出量基本一致,上述各条件下方法均耐用。
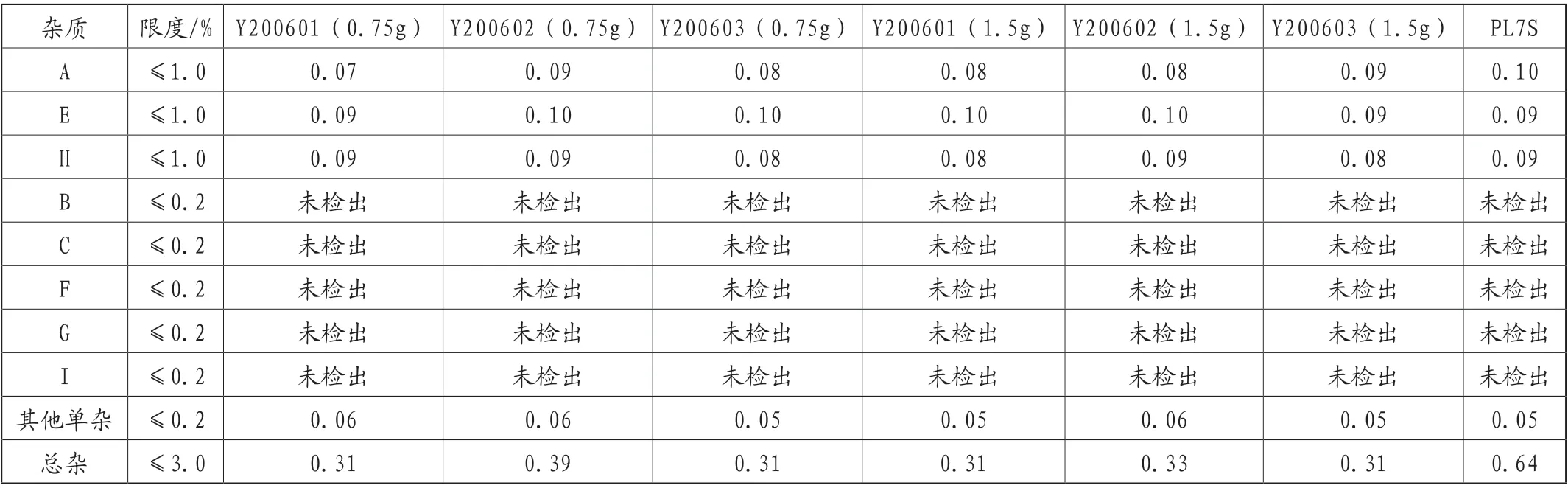
表6 样品中有关物质测定结果
(9)样品测定
采用拟定标准测定6批自制样品和1批参比制剂,均符合规定。结果详见表6。
3.结论
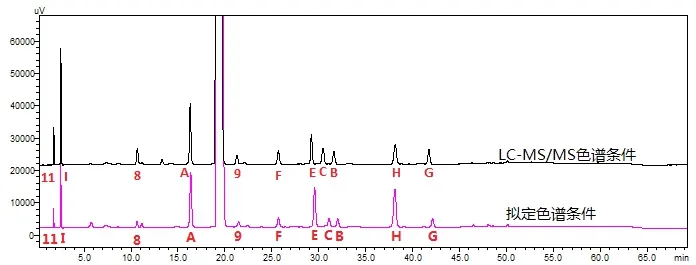
图2 样品与杂质混合溶液在拟定条件、LC-MS/MS条件色谱图

图3 Y200601批加速6月样品拟定条件、LC-MS/MS条件色谱图
根据破坏性试验、影响因素试验和稳定性考察可知杂质A、杂质E、杂质H为降解杂质,其中有一杂质(RRT=0.3)在稳定性考察过程中增加超过0.2%,应对其进行鉴定和安全性评估。注射用头孢呋辛钠进口注册标准中GSK1685761A杂质限度为不大于1.0%,标准中附有其结构式和分子量,此杂质对照品无法获得,多方定制合成失败。根据稳定性结果推测RRT=0.3处的杂质即为GSK1685761A杂质。对此杂质采用LC-MS/MS进行定性研究。将拟定条件中醋酸钠更换为醋酸铵,其他色谱条件不变,作为MS条件。配制样品与杂质混合溶液在拟定条件、上述MS条件下检测,图谱如图2所示,将拟定条件流动相中醋酸钠更换为醋酸铵后,样品及各杂质保留时间基本一致。加速6月样品Y200601批(0.75g,倒置)在拟定条件及MS条件下检测,图谱如图3所示,说明更换缓冲盐,样品中杂质检出一致。因此可在MS条件下对RRT0.3处的杂质进行定性分析。
对Y200601批(0.75g,倒置)加速6月样品中RRT=0.3处杂质进行一级质谱、二级质谱研究。一级质谱解析预测分子量和分子式同GSK1685761A,二级质谱裂解示意图显示其裂解方式符合杂质GSK1685761A的结构特征。因此可以确定拟定条件下RRT=0.3的化合物为GSK1685761A。同进口注册标准,将其限度定为1.0%。
