PRPS1基因突变致综合征性耳聋一家系遗传学分析
2023-02-12任增果秦利涛雷星星郭谦楠娄桂予
任增果,杨 科, 秦利涛, 雷星星, 郭谦楠, 张 冰, 娄桂予, 王 莉
1)河南大学人民医院医学遗传研究所 郑州 450003 2)河南省人民医院医学遗传研究所 郑州 450003 3)郑州大学人民医院医学遗传研究所 郑州 450003
耳聋是严重的致残性疾病,可导致言语交流障碍,影响全球0.1%~0.3%的婴儿[1]。据第2次全国残疾人抽样调查报告,我国听力残疾者有2 780万人,占残疾人总数的33.51%[2]。中国7岁以下的耳聋患儿约有80万,且以每年3万例的速度增长[3]。耳聋的病因复杂,包括遗传因素和环境因素,其中遗传因素约占60%以上。根据是否伴随其他系统症状,遗传性耳聋又可分为综合征性耳聋和非综合征性耳聋,其中综合征性耳聋约占30%。综合征性耳聋除具有耳聋表现外,还存在其他器官或系统的异常,如视网膜或眼底病变、肾功能减退、皮肤色素异常、肌肉骨骼及神经系统异常等。目前已报道的综合征性耳聋有400余种[4],常见的有Waardenburg综合征、鳃耳肾综合征、Pendred综合征、Usher综合征、Alport综合征等。综合征性耳聋具有高度复杂的遗传异质性,临床表现差异大,且尚有一些致病基因未被鉴定,大大增加了临床诊疗难度,因此明确耳聋的分子病因及致病机制,可以为患者及其家属提供准确的遗传咨询和有效的干预手段。本研究通过对一个遗传性耳聋家系进行基因检测,明确其致聋原因,并为家系遗传咨询及高危胎儿的产前诊断提供依据。
1 对象与方法
1.1 研究对象该家系来自河南省人民医院遗传咨询门诊,共收集3代7位成员的相关资料。其中耳聋患者5人,年龄2~56岁,耳聋程度轻到重度不等,家系图见图1。先证者(Ⅲ-2),女,2岁,6个月余发现听力异常、头围小、追视差;Ⅲ-1为男性早产儿,11个月时出现纳差、嗜睡、双耳耳聋、眼球震颤、发育迟缓,后夭折;家系中Ⅰ-2、Ⅱ-2、Ⅱ-3均为先天性耳聋患者,无其他临床表现。家系中耳聋患者均无耳外伤、中耳炎、噪音接触史、耳毒性药物使用史,耳郭、外耳道、鼓膜均未见明显异常,且家系中患者的父母均非近亲结婚,父母孕前期及母亲孕期均未接触致畸或致聋药物。本研究经过河南大学伦理委员会批准和患者及其家属的同意,均签署知情同意书。
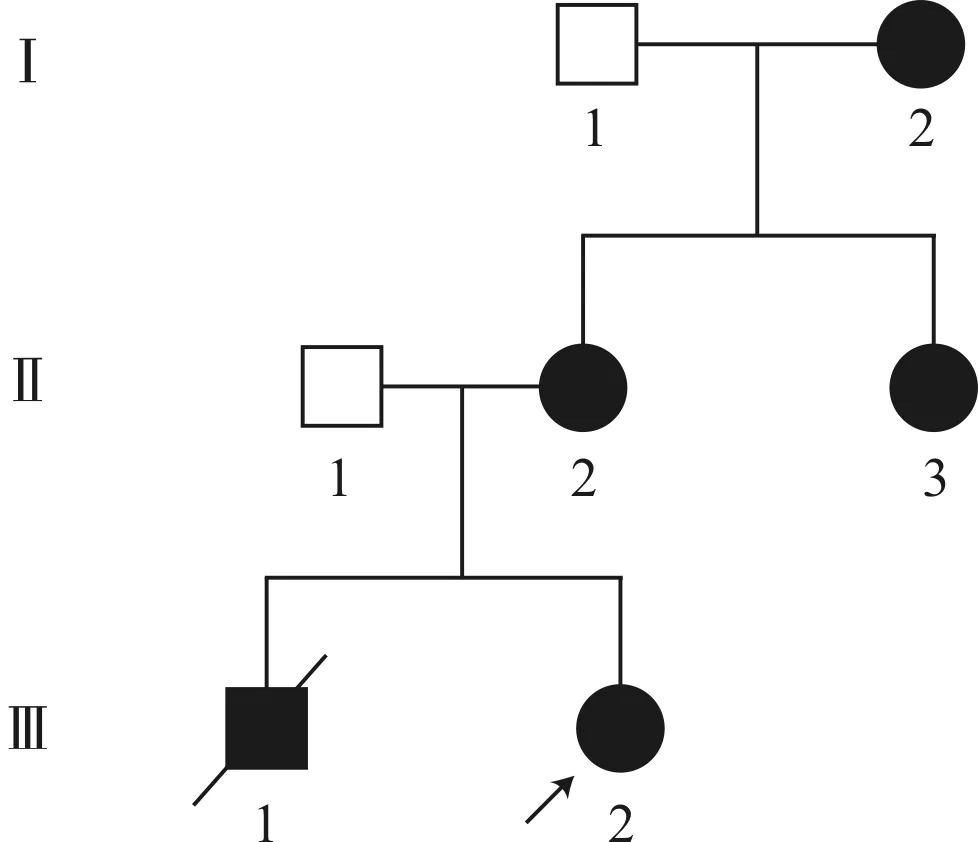
图1 综合征性耳聋家系图
1.2 听力检测小于6岁或不能配合纯音测听检查的耳聋患儿(Ⅲ-2)行听性脑干反应检查,根据文献[5]推荐的矫正值,计算患儿预估听力阈值:预估听力阈值(dB eHL)=听性脑干反应阈值(dB nHL)-矫正值。家系中6岁以上且能配合纯音测听检查的耳聋患者(Ⅰ-2、Ⅱ-2、Ⅱ-3)进行纯音测听检查。按照WHO发布的听力损失分级标准[6],将耳聋分为轻度、中度、中重度、重度、极重度、全聋、单侧聋。
1.3 DNA提取用EDTA-K2抗凝管抽取家系成员外周静脉血各2 mL备用。所有样本均使用北京天根DNA试剂盒,严格按照说明书提取基因组DNA,-20 ℃保存。
1.4 二代测序及数据分析使用Qubit dsDNA HS Assay Kit对先证者DNA进行定量,按照Agilent公司SureSelectXT Target Enrichment System实验流程,采用探针捕获方法对基因组全部外显子区域进行扩增,经Illumina Nextseq500二代测序平台对扩增产物进行测序分析。序列比对参考基因组为UCSC hg19 refGene.txt.gz(http://hgdownload.cse.ucsc.edu/golden Path/hg19/database/)。对检测基因的碱基变异进行筛选,与dbSNP、HapMap和1000Gene数据库进行对比,排除已报道过的非致病性单核苷酸多态性;在人类基因突变数据库(http://www.hgmd.cf.ac.uk)、ClinVar(http://www.ncbi.nlm.nih.gov/clinvar)和相关文献中检索是否为已经明确的致病性突变;并用相关软件(sift、Polyphen2、MutationTaster、REVEL等)对蛋白质功能进行预测。使用UCSC数据库(http://genome.ucsc.edu/)查询变异位置氨基酸的保守性。
1.5 Sanger测序验证根据二代测序结果,对家系相关成员进行验证。从NCBI数据库中获取相关基因的DNA序列,利用Primer 5.0软件针对可疑突变位点设计特异引物,经UCSC数据库in silico PCR验证引物序列的可靠性。PCR法扩增此家系成员该候选位点所在的DNA序列。扩增产物用20 g/L琼脂糖凝胶电泳后,进行双向测序。
1.6 致病性评判根据美国医学遗传学与基因组学学会(the American College of Medical Genetics and Genomics,ACMG)更新的变异解读指南[7],对检测到的基因突变进行致病性评判。
2 结果
2.1 听力检测结果听性脑干反应测听结果显示先证者左耳73 dB nHL,右耳77 dB nHL,矫正后的预估听力阈值为左耳58 dB eHL,右耳62 dB eHL,为双耳中重度耳聋。纯音测听结果提示:Ⅰ-2为双耳轻度耳聋(左耳35.0 dB,右耳29.0 dB);Ⅱ-2为双耳轻度耳聋(左耳34.3 dB,右耳26.5 dB);Ⅱ-3为双耳极重度耳聋(左耳85.8 dB,右耳83.3 dB)。
2.2 基因测序结果先证者X染色体的PRPS1基因(NM_ 002764.3)存在c.46 T>C变异,该突变曾在西班牙家系中报道过[8]。听力异常的Ⅰ-2、Ⅱ-2、Ⅱ-3也存在相同突变,表型正常者(Ⅰ-1、Ⅱ-1)未见该突变。基因测序结果见图2。
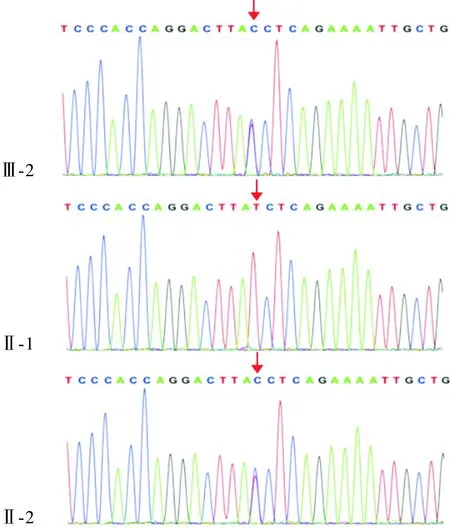
箭头所示为PRPS1基因c.46 T>C突变;Ⅲ-2、Ⅱ-2:突变型;Ⅱ-1:野生型
2.3 致病性评判根据ACMG指南[7],PRPS1基因c.46 T>C为致病性(PS1+PM1+PM2 +PP3+PP1)。PS1:与已确定的致病变异相同,或者有相同的氨基酸改变;PM1:错义突变位于深入研究的无良性变异的外显子功能域;PM2:低频变异;PP3:两种统计方法预测出变异对基因产物有影响;PP1:该家系成员中支持共分离。
3 讨论
随着基因测序技术的快速发展,全外显子组测序已广泛应用于临床,很多疑难、罕见的遗传性疾病得以明确诊断。全外显子组测序即采用高通量测序技术对人类全部外显子区域进行检测分析,因高通量、高效、高准确率等优点,已成为检测遗传性耳聋致病基因的重要手段之一。本研究采用全外显子组测序技术对一个3代耳聋家系进行致病基因检测,发现该家系为PRPS1基因突变所致耳聋,为家系的遗传咨询及后续高危胎儿的产前诊断提供了有力依据。
PRPS1基因位于X染色体长臂,长约36 000 bp,由7个外显子组成,编码磷酸核糖焦磷酸合成酶1(PRS1)。PRS1是核苷酸合成的关键酶,参与嘌呤、吡啶和嘧啶核苷酸的合成以及许多重要的细胞功能[9],如能量代谢和细胞信号转导等[10]。动物实验[11]证明PRPS1基因在小鼠耳蜗和毛细胞中表达,对内耳的发育和维持起着重要作用。
PRPS1基因突变可导致PRS1六聚体晶体结构改变,下游代谢产物对其活性调控发生异常,使PRS1活性异常增高或降低,最终引起疾病发生[12]。活性增高可引起轻重度PRS1超活性(MIM#300661);临床表现可有尿酸增高、痛风,也可伴有其他神经系统症状,如感音神经性耳聋、肌张力下降、共济失调等[13]。活性降低可导致非综合征性耳聋DFN2(MIM#304500)、腓骨肌萎缩症(Charcot-Marie-tooth disease,CMT)-X5(MIM#311070)、Arts综合征(MIM#301835)。有研究[12]表明PRS1活性降低程度与临床表现的严重程度明显相关,其他学者[8]在西班牙家系中也证实了这一观点。DFN2患者的PRS1活性降低程度最轻,患者多表现为先天性或语后进展性感音神经性耳聋;CMT-X5的PRS1活性降低程度较DFN2患者重,除听力异常外,还有视力障碍和周围神经病等表现;Arts综合征的PRS1活性降低程度最重,除上述表现外,还存在早发性肌张力低下、智力低下、运动发育落后、共济失调及感染风险增加等。这3种PRS1活性降低疾病的表型可有一定交叉,其中耳聋为唯一共同的症状,可作为发现此类疾病的关键。PRPS1基因突变所致疾病为X连锁隐性遗传,累及患者多为男性,女性携带者可无症状或出现不同程度的症状。而男性Arts综合征患者多易受感染,尤其是上呼吸道感染,多在5岁之前夭折,故临床中男性Arts综合征患者少见。
本研究测序结果显示先证者的PRPS1基因存在c.46 T>C突变,此变异曾在一西班牙耳聋家系中报道过[8]。该突变靠近蛋白质N端结构域,可导致第16位丝氨酸被脯氨酸取代,此位点在人、猕猴、小鼠、褐家鼠、牛、狼、原鸡等多物种中高度保守。根据ACMG指南[7],该变异为致病性的。有学者[14]在一个英国家系中发现了该基因第16位氨基酸变异。PRPS1基因突变所致疾病具有明显的异质性,同一突变或同一氨基酸位点突变的临床表现不同。已报道的西班牙家系中患者均为女性,同时有Arts综合征及CMT-X5综合征的部分临床表现,如视神经萎缩、视野狭窄、视网膜色素变性、视力损伤等视力异常及听力受损、轻度发育迟缓、肌张力减退、周围神经病变、共济失调等症状。本家系先证者具有Arts综合征的典型表现:感音神经性耳聋、视力异常、发育迟缓等,符合该综合征的临床诊断。Ⅰ-2、Ⅱ-2仅表现为轻度耳聋,Ⅱ-3表现为极重度耳聋,无其他临床表现。经基因测序验证,Ⅰ-2、Ⅱ-2、Ⅲ-3均为PRPS1基因c.46 T>C突变携带者。家系中患者的临床表现差异可能与PRS1活性降低的程度不同有关。本家系中已夭折的Ⅲ-1存在双耳耳聋、眼球震颤、视力异常、发育迟缓、腹胀等症状,虽未能对其进行基因检测验证,但根据其临床表现,推测为Arts综合征。本家系中除Ⅲ-1外,其他患者均为女性,且均有不同程度的症状。该情况可能为X染色体随机失活,亦可能为男性胎儿孕期致死所致。
综上,本研究通过对1个遗传性综合征性耳聋家系进行全外显子组测序,发现其致病原因为PRPS1基因c.46 T>C突变,先证者的临床表现符合Arts综合征的诊断,与PRPS1基因突变所致疾病一致。基因检测为耳聋家系的临床诊断、遗传咨询以及后续的产前诊断提供了有力依据。
