PARP抑制剂调节肿瘤免疫的研究进展
2023-02-12刘洋洋盛誉乔秦志海
刘洋洋,盛誉乔,薛 瑞,秦志海
郑州大学第一附属医院医学研究中心 郑州 450052
多聚ADP核糖聚合酶[poly(ADP-ribose)polymerase,PARP]超家族由17种具有不同功能的蛋白质组成,负责将 ADP-核糖基团转移到靶蛋白以启动 ADP-核糖基化,这是所有生物体中高度保守的翻译后修饰。PARP家族参与多种细胞功能的调节,包括染色质结构、转录、复制、重组和DNA修复。依据结构和功能的不同,PARP家族成员包括DNA依赖型、端锚聚合酶型、Cys-Cys-Cys-His型和macroPARP型等4种类型[1]。DNA依赖型包括PARP1、PARP2、PARP3,主要参与DNA损伤修复过程,其中PARP1承担了90%的功能,在DNA碱基切除修复和单链断裂修复过程中发挥重要作用[2]。PARP抑制剂主要是通过抑制PARP1和PARP2的催化活性和将PARP1或PARP2“捕获”于损伤的DNA上发挥作用[3]。迄今为止,全球共有4种PARP抑制剂上市,分别是奥拉帕利(Olaparib)、鲁卡帕利(Rucatinib)、他拉唑帕利(Talazoparib)和尼拉帕利(Niraparib)。2014年,美国食品药品监督管理局(FDA)批准Olaparib用于治疗种系BRCA突变晚期的卵巢癌[4]。最近,Niraparib已被证明可显著延长卵巢癌患者的无进展生存期,并因此获得美国食品药品监督管理局批准用于治疗铂类敏感性卵巢癌复发[5]。除卵巢癌外,PARP抑制剂在乳腺癌治疗中也显示出巨大潜力[6],已有多项临床试验正在评估PARP抑制剂对于晚期乳腺癌的疗效[7]。本文依据最新的研究数据,对PARP抑制剂在各类免疫细胞调节过程中的作用以及PARP抑制剂在临床应用中的毒副作用进行了总结。
1 PARP抑制剂的作用机制
最初人们认为PARP抑制剂通过“合成致死”的原理杀死肿瘤细胞[8]。这一原理指出两种非致死性突变中的任何一种单独存在不影响细胞活力,但是两种突变同时存在则可导致细胞死亡。PARP1可与DNA损伤位点相结合(大多为单链DNA断裂) ,通过催化多聚ADP核糖链在蛋白底物上的合成,募集其他DNA修复蛋白到损伤位点,共同修复DNA损伤。PARP抑制剂通过与PARP1或PARP2催化位点结合,将PARP蛋白“捕获”在DNA损伤位点上且无法脱落,从而使得DNA修复蛋白无法结合损伤位点,导致DNA复制叉停滞和DNA复制无法顺利进行,细胞将启动同源重复修复(homologous recombination repair,HRR)来修复DNA损伤,若HRR功能受损,DNA无法修复,则最终导致细胞死亡[9]。BRCA1/2和其他“类BRCA”蛋白在HRR中起到重要作用,研究[10]发现携带突变BRCA的肿瘤细胞对PARP抑制剂的敏感度是携带野生型BRCA肿瘤细胞的1 000倍,因此推测其他原因导致的HRR缺陷也可能使肿瘤细胞对PARP抑制剂敏感。这一重大发现推进了PARP抑制剂作为单一疗法在临床上的应用。有研究[11-12]结果显示PARP抑制剂能够影响抗肿瘤免疫反应,其与癌症相关免疫之间的串扰可能提升抗癌效果。
2 PARP及其抑制剂对抗肿瘤免疫的影响
2.1 对T细胞的影响T细胞是机体免疫系统中对抗癌细胞的主力军。PARP1和 PARP2蛋白均在胸腺细胞中表达,但只有PARP2在胸腺细胞发育中发挥重要作用,PARP2敲除小鼠体内CD4+CD8+胸腺细胞减少,可能与PARP2抑制DNA双链断裂积累诱导的细胞凋亡有关[13]。在PARP1缺陷小鼠中特异性敲除T 细胞的PARP2会破坏T细胞稳态,CD4+和CD8+T细胞减少,其中CD8+T细胞减少更为明显,同时DNA损伤和细胞凋亡标志物表达增强,提示T细胞的减少可能是基因错配的累积导致的细胞死亡,而不单单是成熟障碍所致[14]。与T细胞PARP1或PARP2单缺陷相比,PARP1/2双重缺陷小鼠乳腺癌细胞增殖更快,且肿瘤组织内CD4+和CD8+T细胞浸润减少[10,15]。
PARP在T细胞分化中也起着重要作用。PARP1能够通过活化T细胞的核因子NFAT影响CD4+T细胞的分化[16]。PARP1缺乏可导致依赖NFAT的细胞因子(如IL-2和IL-4)表达降低,进一步影响免疫细胞的分化[17]。PARP1缺失或抑制可能会使CD4+T细胞偏向1型T辅助细胞(Th1)分化[18]。PARP1可调节CD4+T细胞中转化生长因子β(TGF-β)受体的表达,在PARP1缺失的情况下持续给予TGF-β1和IL-6刺激,CD4+T细胞在体外分化为Th17的能力增强[16]。在哮喘小鼠模型中,Olaparib可增加Th1相关细胞因子如干扰素(IFNγ)和转录因子T-bet的表达,同时抑制Th2相关细胞因子如IL-4、IL-5、IL-6、IL-13和巨噬细胞集落刺激因子(M-CSF)的表达[15]。而在关节炎小鼠模型中,PARP抑制与Th1相关细胞因子肿瘤坏死因子α(TNFα)和IFNγ的表达降低有关,并可抑制部分Th1细胞的增殖[19]。
除了影响T细胞分化外,PARP1和PARP2还影响T细胞功能。在BRCA缺陷卵巢癌、三阴性乳腺癌及小细胞肺癌小鼠模型中,Olaparib可能通过激活cGAS-STING通路上调CXCL10及CCL5表达,招募CD8+T细胞至肿瘤组织,激活抗肿瘤免疫反应[11-12,20]。Olaparib还可降低T细胞免疫检查点受体PD-1、Tim-3和Lag-3的表达,这些受体与T细胞抑制和衰竭有关[11]。叉头样转录因子3(Foxp3)是控制调节性T细胞(Treg)发育和免疫抑制功能的关键转录因子之一[21]。研究[22]显示,PARP1可与Foxp3相互作用并诱导其多聚ADP-核糖化。T细胞单一缺陷PARP1或PARP2的模型小鼠外周淋巴组织中T细胞数量没有变化[4],但PARP1和PARP2双重缺陷会导致乳腺癌模型小鼠外周CD4+CD8+T细胞减少[16]。上述结果表明,PARP抑制剂与效应T细胞的激活有关。
2.2 对B细胞的影响B细胞在肿瘤免疫中扮演重要角色,其促肿瘤或者抗肿瘤效应取决于肿瘤微环境[23]。PARP能够影响B细胞稳态。PARP1/2双重缺陷虽然不影响小鼠骨髓B祖细胞和周围成熟B细胞数量,但会使外周血中过渡性和滤泡性B细胞减少,提示 PARP在B细胞分化过程中可能发挥重要作用[24]。免疫球蛋白V(D)J基因重排对于前B细胞生成免疫球蛋白至关重要。V(D)J重排导致的DNA双链断裂可由PARP1介导的NHEJ途径修复[25]。B细胞PARP1/2双重缺陷并不影响V(D)J重排,但会导致血清IgG对非T细胞抗原的应答水平降低[26]。PARP还可抑制B细胞在生发中心的分化与成熟[27]。但目前尚不清楚在实体恶性肿瘤中PARP抑制剂如何影响B细胞稳态和免疫球蛋白的产生。
2.3 对NK细胞的影响NK细胞能够在抗原暴露前杀死肿瘤细胞,在抗肿瘤免疫应答中起关键作用。肿瘤坏死因子相关凋亡诱导配体(TRAIL)是NK细胞的关键效应分子,已被证明可以阻止肿瘤的发生、生长和转移[26]。Olaparib 和Veliparib能够上调多种癌细胞(白血病、卵巢癌和肺癌细胞等)表面促凋亡分子Fas及死亡受体5(DR5)的表达,使得癌细胞对TRAIL诱导的细胞凋亡更加敏感[28]。Olaparib可能通过上调BRCA野生型或突变型前列腺癌细胞表面死亡受体TRAIL-R2的表达,增强其对NK细胞的敏感性(如图1)[29]。在人急性髓细胞性白血病移植小鼠模型中,敲除或抑制PARP1可诱导耐药白血病干细胞上NKG2D配体的表达,该配体是NK细胞抗肿瘤免疫的关键介质,继而促使NK细胞定点清除白血病干细胞并抑制白血病的发生[26,30]。这些证据表明PARP抑制剂在NK细胞介导的抗肿瘤免疫中占有一席之地。
2.4 对树突状细胞(DC)的影响DC是肿瘤微环境中关键的抗原呈递细胞,能够激活并诱导T细胞分化[27,31]。Olaparib能够显著增加成熟DC的比例并增强其抗原递呈能力[12]。在BRCA缺陷的卵巢癌和三阴性乳腺癌小鼠模型中,Olaparib能够激活肿瘤细胞cGAS/STING通路,从而激活DC中TBK1/IRF3信号(如图1)[6-7,11-12]。此外,与单PARP1缺陷、单PARP2缺陷或正常对照小鼠相比,PARP1/2双重缺陷的乳腺癌小鼠癌组织中CD11b+DC百分比更高[11,16]。但上述结论皆为PARP及其抑制剂对DC募集和功能的间接影响,目前尚未有其直接影响DC功能的证据。有趣的是,在自身免疫性脑脊髓膜炎小鼠模型中,PARPi能够直接抑制小鼠骨髓源性DC的NF-κB活化以及成熟,继而影响DC的迁移和抗原递呈功能,降低疾病严重程度和复发率[28,32]。
2.5 对肿瘤相关巨噬细胞(TAM)的影响TAM是构成肿瘤微环境的重要免疫细胞。巨噬细胞具有高度异质性,在被招募、迁移至肿瘤间质后,可以通过改变自身的免疫表型来适应肿瘤微环境,包括促肿瘤表型M2型和抗肿瘤表型M1型[33]。M1型的主要作用是驱动Th1反应,具有对微生物和肿瘤细胞的细胞毒性。M2型(又称替代激活的巨噬细胞)可被IL-4或M-CSF激活,生成精氨酸酶-1,将精氨酸分解为鸟氨酸,促进细胞外基质的形成,促进肿瘤的侵袭和转移。M2型巨噬细胞还可通过分泌IL-10等抗炎因子抑制炎症,通过分泌血管生成相关因子参与肿瘤血管的生成和细胞外基质的重塑,通过下调M1型巨噬细胞介导的功能和适应性免疫来控制炎症反应,发挥促肿瘤作用[34]。巨噬细胞具有吞噬肿瘤细胞并呈递肿瘤特异性抗原以诱导适应性抗肿瘤免疫的潜力[35]。靶向TAM已成为潜在的肿瘤免疫治疗新策略。
巨噬细胞的代谢途径与其表型和功能密切相关,M1型主要表现为糖酵解增强、谷胱甘肽水平升高,M2型脂肪酸氧化水平增强[36-37]。巨噬细胞的特性使它们容易受到多种压力和损伤的影响。PARP1可以保护巨噬细胞免于氧化诱导的死亡[38],可以通过与MLL1甲基转移酶的相互作用抑制IL-6的转录[39-40]。在BRCA缺陷的乳腺癌中,PARP抑制剂通过影响TAM的代谢,进而影响其免疫抑制程度[41]。TAM可能会抑制PARP抑制剂诱导的肿瘤细胞DNA损伤,从而减弱sting依赖的抗肿瘤免疫[42](如图1)。但是,目前关于PARP抑制剂对巨噬细胞直接作用的报道较少,作用机制也不清楚。
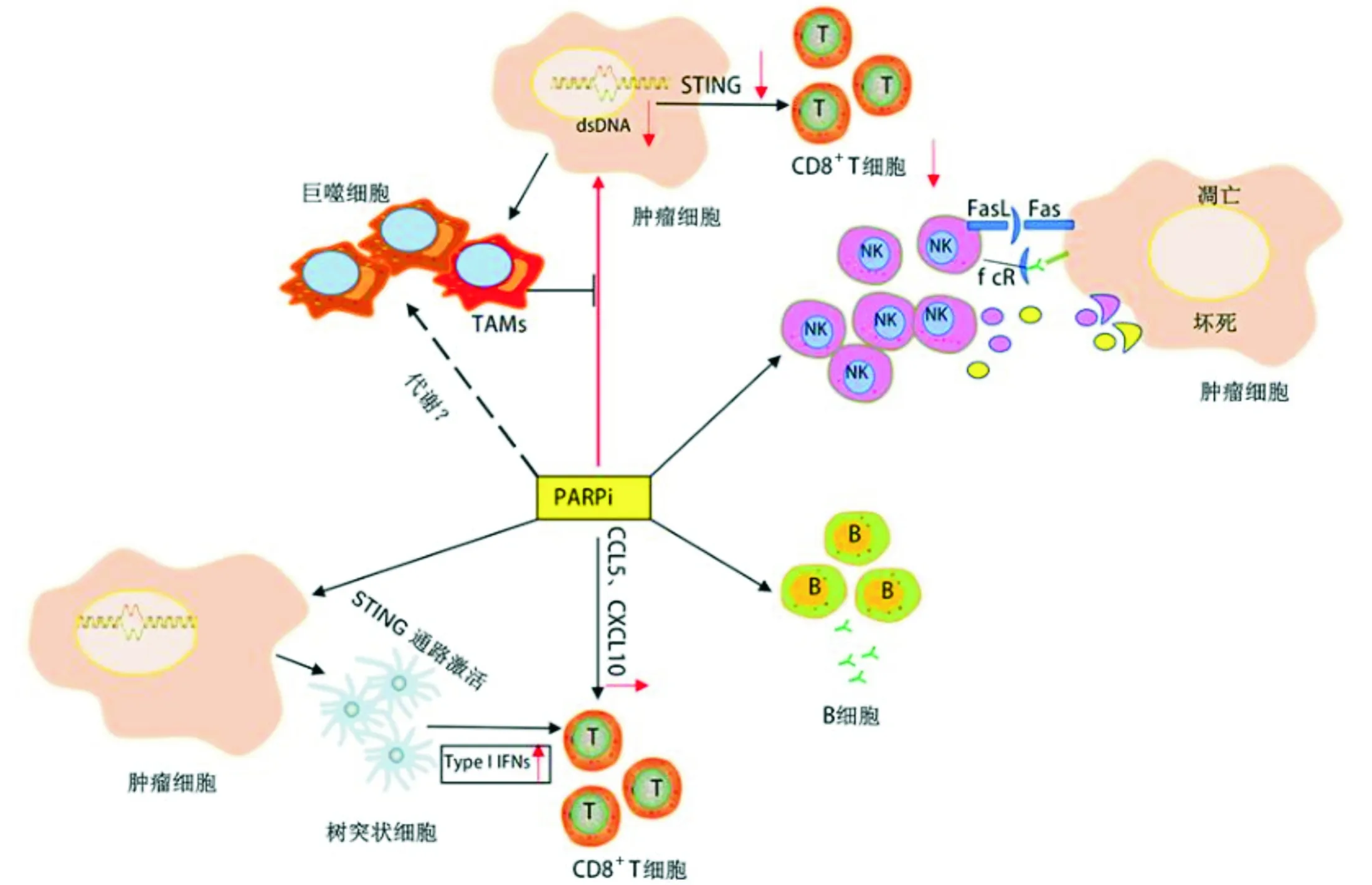
图1 PARP抑制剂对于各类免疫细胞的调节作用以及对肿瘤细胞的影响
3 PARP抑制剂的主要毒副作用
3.1 血液系统不良反应除了修复DNA,PARP1还参与了骨髓和血液系统细胞的分化,以及血小板的生成;PARP2也被证实在红系细胞分化中发挥重要作用[43]。PARP抑制剂大部分的3或4级不良反应为血液学不良反应,主要包括贫血、血小板减少、中性粒细胞减少等,也是导致减量、中断和停止用药的最主要原因。贫血是PARP抑制剂最常见的不良反应,可能与药物靶向PARP2有关。在3期维持试验中,接受Olaparib、Niraparib和Rucatinib治疗的卵巢癌患者贫血发生率分别为44%、50%和37%,3或4 级不良反应发生率分别为19%、19%和25%[6,44-45];血小板减少发生率为14%、61%和27%,3或4级血小板减少的发生率为1%、34%和5%,常发生在治疗的第一个月[44,46]。血小板计数是Niraparib剂量调整的重要指标[47]。中性粒细胞减少总发生率为18%~30%,其中4%~20%为3或4级不良反应[45]。总之,临床推荐PARP抑制剂治疗的第1个月应每周监测全血细胞计数,治疗第1年为每月监测,之后进行定期监测。如因发生3或4级血液系统不良反应导致治疗暂停,在恢复用药后,应每周监测全血细胞计数,直到恢复至正常水平。
3.2 消化系统不良反应恶心是最常见的消化系统不良反应,发生率为70%~76%,其他常见胃肠道不良反应包括便秘、呕吐和腹泻,1或2级不良反应多见,仅3%~4%的患者发生3或4级不良反应[44,46]。为了防止消化系统不良反应,可预防性使用止吐药物,如5-羟色胺拮抗剂。如果患者出现突发性恶心、呕吐症状,可以依次添加不同类别的止吐药,包括甲氧氯普胺、地塞米松等[48]。持续恶心需要接受止吐药物治疗,如果影响正常生活和(或)体重降低超过5%,在排除其他原因后,需降低PARP抑制剂剂量[49]。
3.3 肾毒性PARP抑制剂的肾毒性主要表现为血肌酐升高。根据临床数据,使用Rucatinib的患者血肌酐升高发生率为15%[44],Olaparib为11%[45],Niraparib未见相关报道。PARP抑制剂可能是通过抑制MATE1和MATE2-K肾转运蛋白,使肌酐分泌异常增强,导致肾毒性[44]。因此,PARP抑制剂用药初始即应检查患者电解质水平,在治疗过程中,肾小球滤过率正常时可不减量继续治疗,若肌酐水平升高且肾小球滤过率下降,则需中断治疗,并评估肾功能损伤程度[50]。
3.4 其他不良反应疲劳发生率为59%~69%,大部分症状为1或2级[44,46]。实验室指标常见异常为胆固醇和血清转氨酶升高[51]。不常见的毒副作用包括神经毒性、呼吸道毒性、肌肉骨骼疼痛、皮肤毒性、心血管毒性等,大多数症状并不严重,对症处理可缓解。
4 展望
从发现PARP抑制剂与BRCA的合成致死机制到PARP抑制剂获得批准,共历经了十余年。如今,PARP抑制剂仍然是一个热门的研究领域,多项临床试验正在进行,主要用于BRCA突变的卵巢癌和HER2阴性转移性乳腺癌的治疗。肿瘤细胞可能通过多种机制对PARP抑制剂产生抗药性,原因包括BRCA基因突变使HRR缺陷得到恢复、PARP基因突变导致PARP蛋白缺失、PD-L1表达上调[52]。Veliparib联合化疗治疗高分化浆液性卵巢癌获得了较为满意的临床缓解[53],但是两者联合产生的骨髓抑制也是需要解决的难题。Olaparib引起的DNA损伤扩增可增加肿瘤突变负荷,进而通过上调PD-L1增强肿瘤免疫原性。临床试验中Olaparib、Niraparib与抗PD-L1单抗的联用也获得了良好的抗癌效果[54-55]。未来研究的重点将是通过联合用药方案,使PARP抑制剂产生更大价值和疗效,并尽可能地削弱PARP抑制剂联合治疗的毒副作用。
