PARK2介导的线粒体自噬对锰诱导的神经细胞毒性的作用
2023-02-06胡宏涛蒋智钢周远忠范奇元
张 玥,胡宏涛,赵 颖,蒋智钢,周远忠,范奇元,2
(1.遵义医科大学 公共卫生学院,贵州 遵义 563099;2.遵义医药高等专科学校 卫生管理系,贵州 遵义 563006)
锰是氨基酸、脂质、蛋白质和化合物代谢所必需的微量元素[1],与发育、新陈代谢和抗氧化密切相关[2-4]。锰摄入不足将造成生殖缺陷、体重减轻、生长障碍、皮肤疾病和心情改变等[5],接触过量锰则会导致锰中毒,以中枢神经系统受损为主要表现[6]。锰对神经系统的损害,多以多巴胺能神经细胞为靶细胞,可导致该细胞中线粒体受损。维持多巴胺能神经细胞的正常功能,依赖于细胞中线粒体的质量和数量[7],线粒体参与细胞中多种功能的正常发挥:能量转换、钙稳态、细胞凋亡和代谢合成等[8]。细胞内活性氧(ROS)产生过多超过了细胞自身的清除能力、ATP产生受阻、线粒体DNA损伤、半胱天冬酶释放和电子传递复合物酶缺陷等[9]都将诱导线粒体功能失调,产生神经细胞中毒效应[10]。
线粒体自噬可以清除受损或不完整的线粒体,被认为是控制线粒体数量和质量的一种重要机制[11],参与维持自身功能的完整性,是细胞内线粒体新陈代谢的主要途径,对维持细胞的稳态具有重要意义[12]。已有的研究已经证实线粒体受损诱导蛋白激酶 PINK1聚集到线粒体外膜,其下游蛋白 Parkin泛素化线粒体外膜上的蛋白,介导线粒体自噬的发生[13-15]。虽然锰与线粒体自噬之间的关系已有相关研究报道,但PARK2介导的线粒体自噬如何影响锰对多巴胺神经元细胞毒性未见相关报道。本课题组前期工作[16-19]发现锰中毒伴随PARK2基因表达下降:锰暴露工人血液和唾液中PARK2基因下调;锰暴露大鼠血、 大脑中PARK2基因下调; 大鼠在脱离锰接触后,血中PARK2基因的下降恢复正常, 而PARK2基因的产物蛋白Parkin在中脑、纹状体的表达也恢复正常, 伴随多巴胺神经元的恢复; 在SH-SY5Y细胞中, 染锰导致PARK2基因和Parkin蛋白的下调与线粒体损伤和多巴胺的分泌下降有关。但PARK2/Parkin的下调如何影响锰的毒性不明。因此构建PARK2基因过表达/敲低的细胞有助于进一步研究PARK2基因/Parkin蛋白在多巴胺能神经细胞染锰时的线粒体自噬调节,这对于探讨锰中毒作用机制有重要意义。
1 材料与方法
1.1 材料 氯化锰(MnCl2·4H2O )购于生工生物工程( 上海) 股份有限公司;PARK2过表达和敲低质粒购于中洪博元生物技术有限公司(南昌); LipotamineTM2000 购自中洪博元生物技术有限公司(南昌);多巴胺 (Dopamine,DA) ELISA试剂盒(上海生工生物工程股份有限公司);Parkin单克隆抗体(美国CST,#4211);LC3单克隆抗体(美国Abcam,ab192890);OPTN单克隆抗体(美国Abcam,ab213556);P62单克隆抗体(美国Abcam,ab109012);GAPDH单克隆抗体(上海碧云天生物技术研究所)。
1.2 细胞培养 SK-N-SH细胞购于上海和元生物公司,培养于含有10%胎牛血清(FBS)及1%双抗的MEM培养基中,置于37 ℃、5%CO2培养箱内孵育,每隔1天更换培养基。为更好观察PARK2对锰引起的自噬, 实验中使用2% FBS MEM培养基。
1.3 细胞转染 使用中洪博元生物技术有限公司构建的PARK2过表达和敲低质粒来转染细胞。取8.75 μL LipotamineTM2000和2.5 μg目的质粒溶于150 μL Opti-MEM无血清培养基中,混匀室温下孵育30 min。弃原培养基,清洗细胞后,每孔加入1.7 mL基础培养基,再将混匀的质粒溶液逐滴加入。置于37 ℃孵箱内培养4 h,弃培养基,每孔加入2 mL基础培养基培养24 h。加入嘌呤霉素进行稳转株的筛选,得到稳转株细胞。稳转细胞株用PARK2 mRNA 和Parkin蛋白的表达来验证。
1.4 qPCR 用Trizol法提取Total RNA后用PrimeScript RT reagent Kit(Takara,Japan)试剂盒逆转录为cDNA,再用SYBR®Premix Ex Tag(Takara,Japan)进行qPCR扩增。实验以GAPDH为内参,采用相对定量法(Q = 2-ΔΔCt)计算目的基因的表达量。qPCR 探针序列见表1。

表1 qPCR 探针序列表
1.5 Western blot实验 用RIPA、PMSF和蛋白酶抑制剂提取SK-N-SH细胞的总蛋白,再用BCA蛋白检测试剂盒检测总蛋白浓度 。用十二烷基硫酸钠/聚丙烯酰胺凝胶电泳法分离等量的蛋白质(20 μg),并将蛋白转移到聚亚甲基氟化物膜。在5%脱脂牛奶中封闭2 h,在4 ℃下孵育一抗过夜,洗涤后用辣根过氧化物酶标记的二抗 (1∶10 000) 孵育1 h。最后用凝胶图像分析系统计算目的蛋白相对含量=目的蛋白灰度值/内参GAPDH蛋白灰度值。
1.6 细胞分组及技术路线 慢病毒构建PARK2过表达/敲低稳转株后,分为空载质粒组、PARK2过表达组、PARK2敲低组3组,对其染毒24 h后进行后续实验。技术路线见图1。
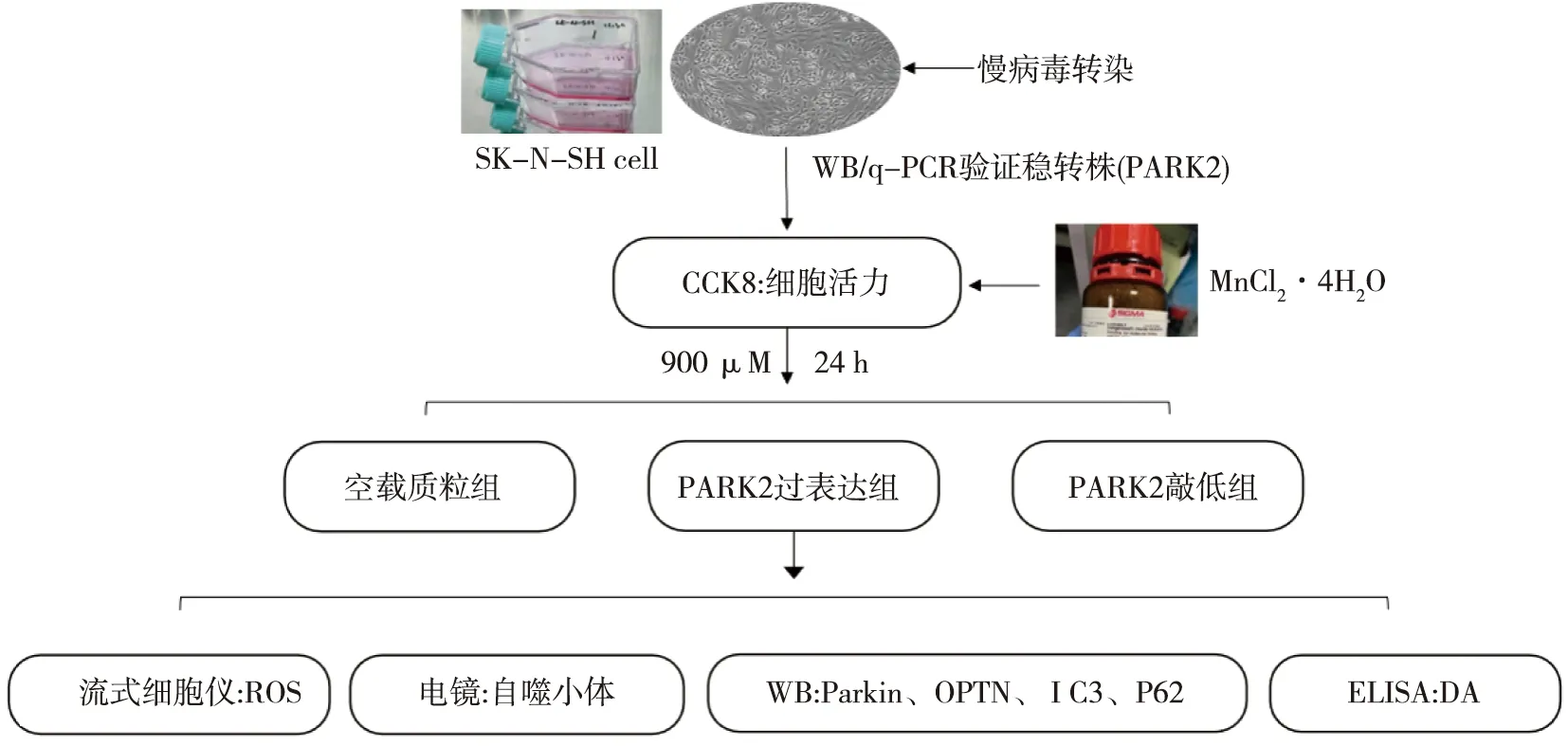
图1 技术路线
1.7 CCK 8法测锰对细胞增殖-毒性 在细胞对数期收集细胞,以1×105个/mL的密度接种于96孔板培养24 h后加入配制好的含不同Mn2+浓度(0~3 000 μM)的2% FBS培养基培养24 h,然后每孔加入10 μL CCK8工作液(避光操作),2 h后用酶标仪检测在波长450 nm处的吸光度。根据吸光度值算出细胞活力。
1.8 荧光探针测ROS水平 将20 μL H2DCFDA储存液加入到10 mL Assay Buffer中,配置成染色液 (终浓度10 μM)。将1mL染色液覆盖细胞于培养箱内孵育1 h后用PBS洗涤细胞,再消化、离心、重悬细胞后用流式细胞仪检测。
1.9 透射电镜下观察自噬小体数量 细胞染Mn2+24 h后用2.5%戊二醛溶液和1%锇酸固定液固定,再脱水包埋固化后切片染色,用透射电镜观察、拍片。
1.10 ELISA法测DA含量 按多巴胺 ELISA试剂盒说明书进行:将细胞培养上清移离心, 在标准孔、空白孔和样本孔中分别各加入50 μL标准品稀释液、检测缓冲液和细胞上清液,然后加入50 μL生物素标记多巴胺抗体工作液,于37 ℃孵育45 min。洗涤后每孔加入 100 μL 稀释的辣根过氧化物酶标记的链霉亲和素(1∶100 稀释),封板后于37 ℃孵育30 min。洗涤后每孔加入90 μL显色底物TMB,避光室温孵育 15 min后用酶标仪450 nm波长测定OD值。
2 结果
2.1 WB和q-PCR实验验证PARK2 基因过表达/敲低稳转株 图2A显示, 与空细胞组比较,空载质粒组无蛋白表达,PARK2 过表达组中Parkin蛋白表达,表明PARK2 过表达稳转株构建成功。图2B显示,与空细胞组比较,PARK2基因的表达在空载质粒组无差别,而在PARK2 敲低组中PARK2 基因的表达量仅为空白组的9.17%(P<0.05),表明PARK2 基因敲低稳转株构建成功。
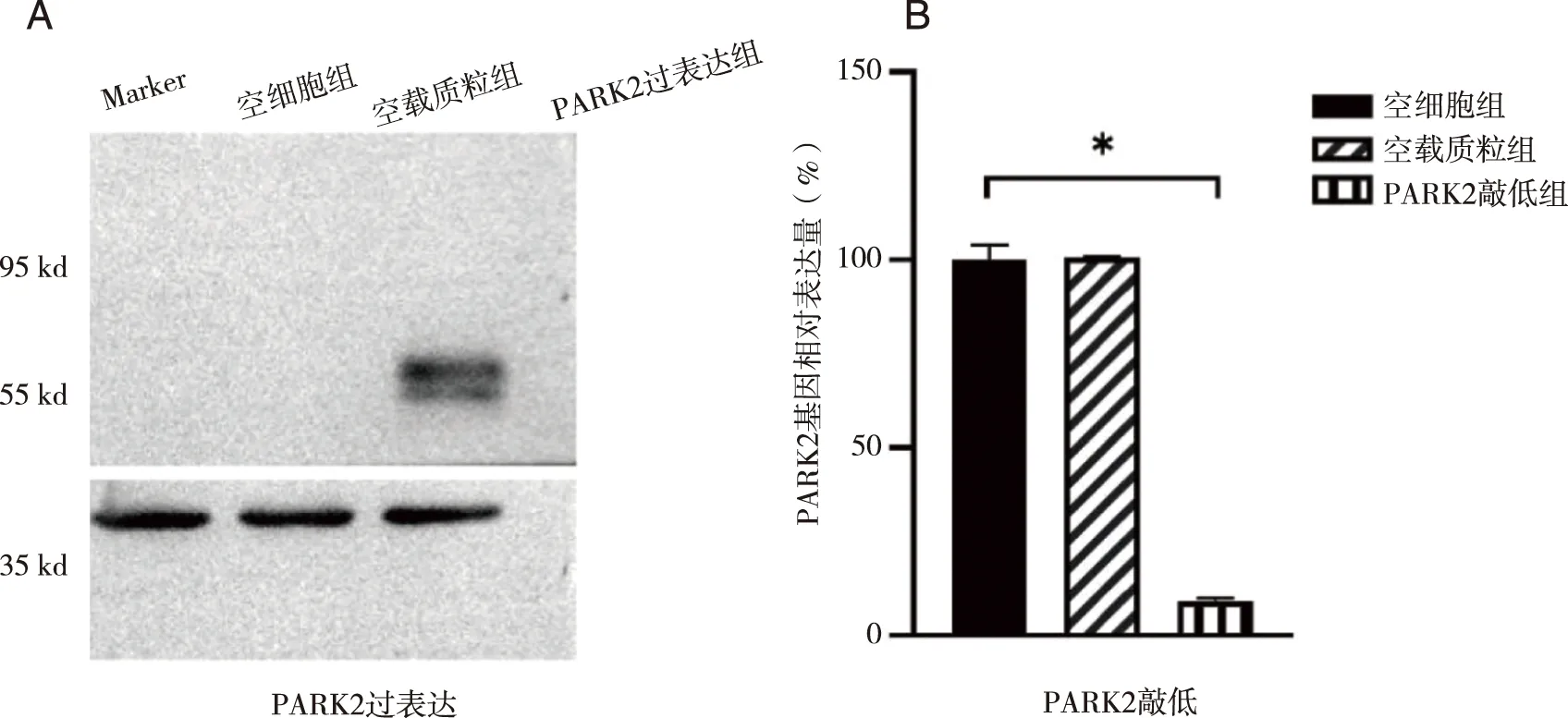
A:PARK2过表达后各组细胞内Parkin蛋白表达 (55KD);B:PARK2敲低后各组细胞内PARK2基因相对表达量; *:与空细胞组比较,图2 WB/q-PCR验证PARK2过表达/敲低稳转株
2.2 不同浓度Mn2+对SK-N-SH细胞增殖毒性 为更好观察自噬,采用血清饥饿法。 图3A显示2%FBS培养基不影响细胞活力, 故2%FBS 培养基用来评价锰的毒性。 设置含不同浓度Mn2+(0~3 000 μM)的培养基培养细胞24 h后,用CCK 8法测细胞活力。图3B显示,与对照组比较,125、250和500 μM Mn2+组存活率无差异(P>0.05),其余各组的细胞存活率随Mn2+浓度而较低(P<0.05),选择900 μM作为染锰剂量进行后续实验。
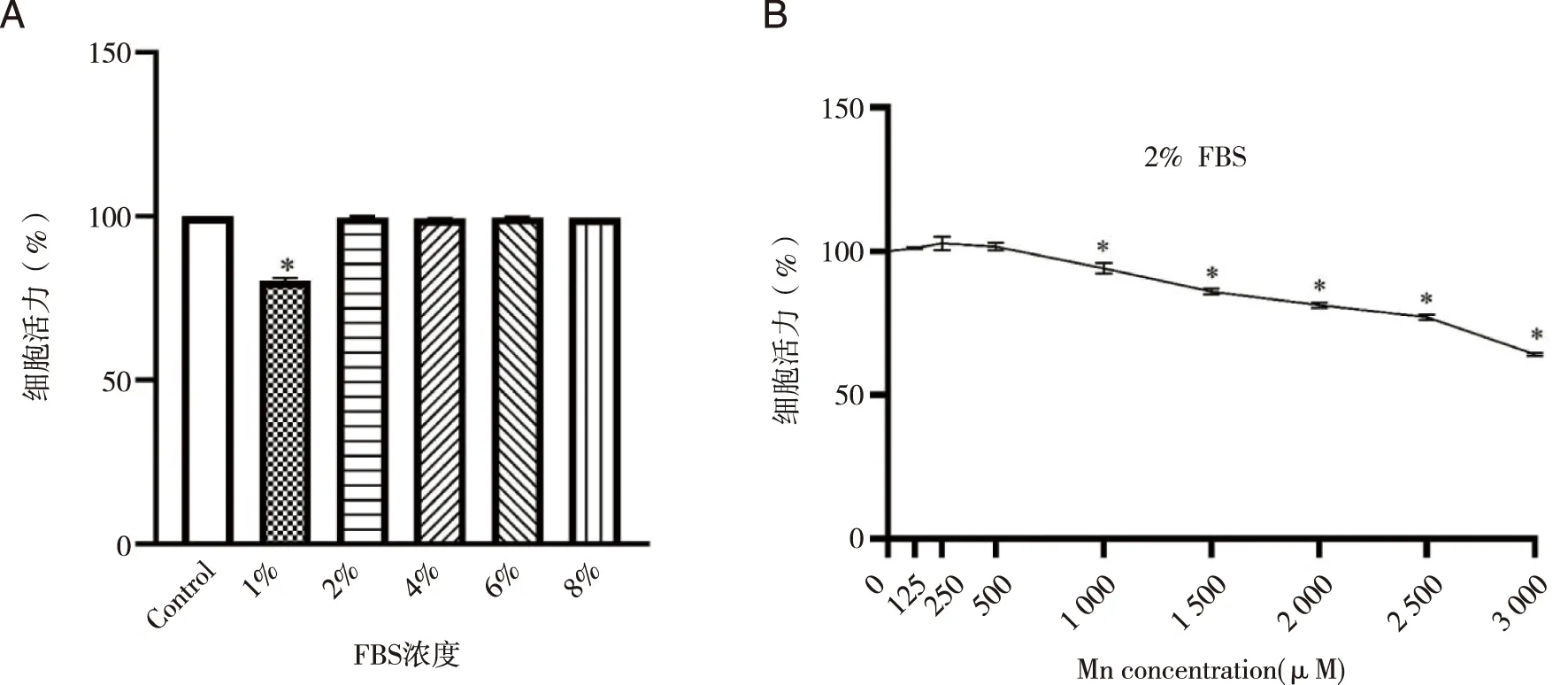
*:与对照组比较,图3 不同浓度Mn2+对细胞的增殖毒性
2.3 荧光探针测细胞内ROS水平 用流式细胞仪检测细胞内ROS生成水平(见图4)。与空载质粒组比较,PARK2过表达组ROS含量较低(P<0.05)。
2.4 透射电镜观察自噬小体数量 在透射电镜下观察不同处理组细胞内自噬小体数量,见图5。细胞内可见多个内含物质的囊泡结构,即为自噬小体(箭头)。与空载组比较,PARK2过表达组自噬小体数量较多(P<0.05)。
2.5 WB检测OPTN、P62和LC3Ⅱ/Ⅰ蛋白表达 经WB法检测了不同处理组细胞内OPTN、P62和LC3Ⅱ/Ⅰ 蛋白表达情况(见图6)。与空载组比较,PARK2过表达组OPTN蛋白表达量较低(P<0.05),PARK2敲低组OPTN蛋白表达量较高(P<0.05);PARK2敲低组P62蛋白含量较高(P<0.05);PARK2过表达组LC3Ⅱ/Ⅰ较高(P<0.05)。
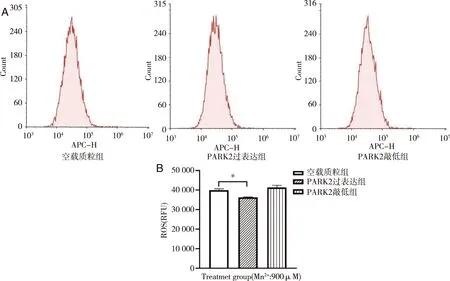
A:流式细胞仪检测结果;B:统计结果;*:与空载质粒组比较,图4 不同处理组细胞内ROS水平
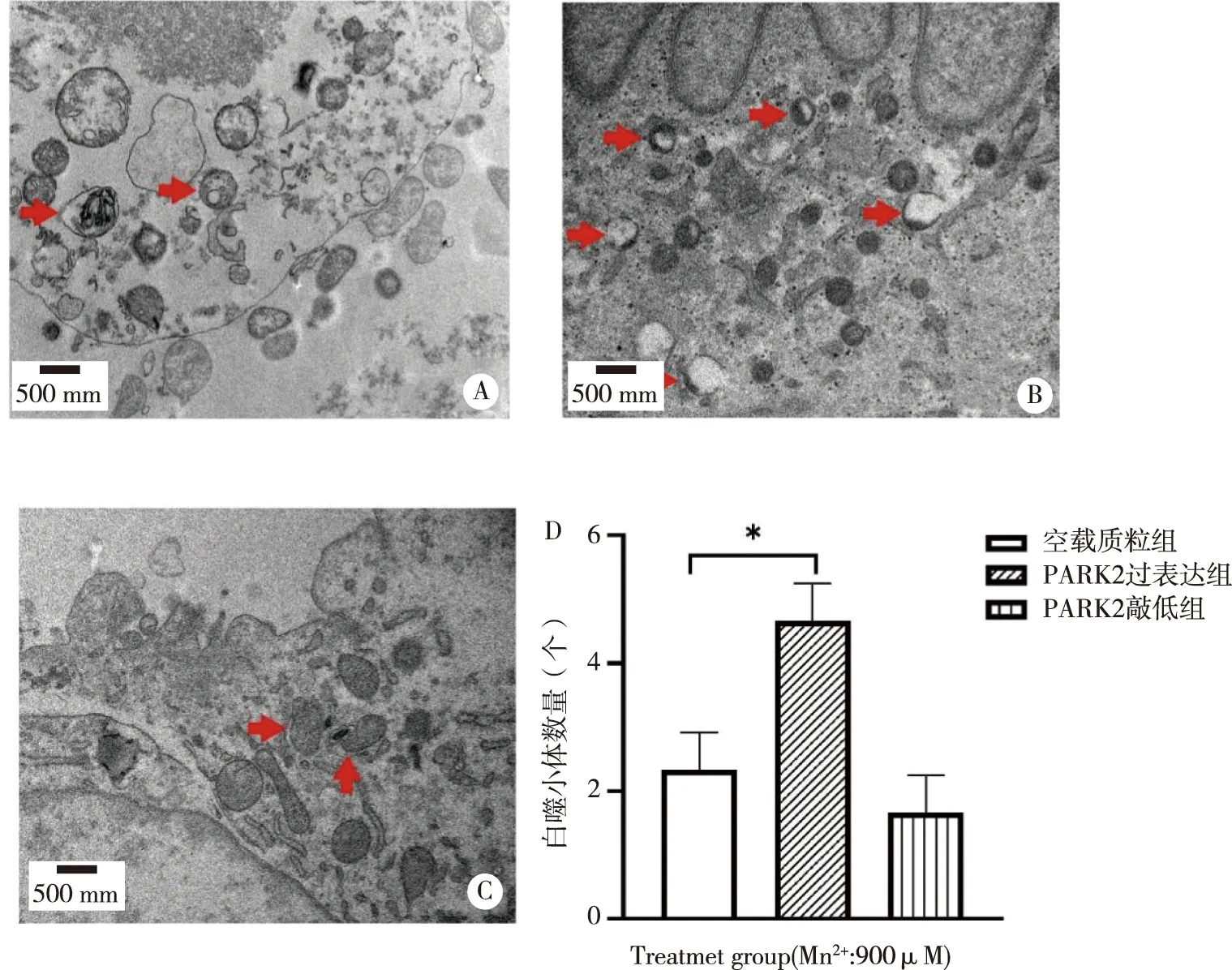
A、B、C:空载质组、PARK2过表达组、PARK2敲低组电镜下自噬小体;D:统计结果;箭头:自噬小体;*:与空载质粒组比较,图5 电镜下观察各组细胞内自噬小体数量
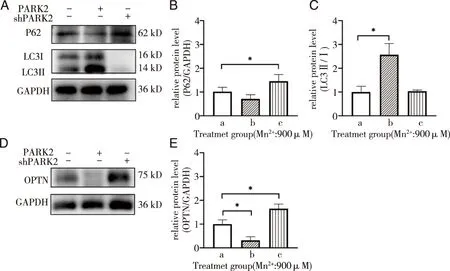
A、D:P62/LC3Ⅱ/Ⅰ/OPTN WB代表性条带图;B、C、E:P62/LC3Ⅱ/Ⅰ/OPTN蛋白表达量统计结果;*:与空载质粒组比较,P<0.05;a、b、c:空载质粒组、Parkin过表达组、Parkin敲低组;图6 不同处理组细胞内OPTN、P62和LC3Ⅱ/Ⅰ蛋白的表达
2.6 ELISA法检测多巴胺(DA) 用ELISA法检测细胞培养基中DA水平(见图7)。与空载组比较,PARK2过表达组DA水平较高(P<0.05)。
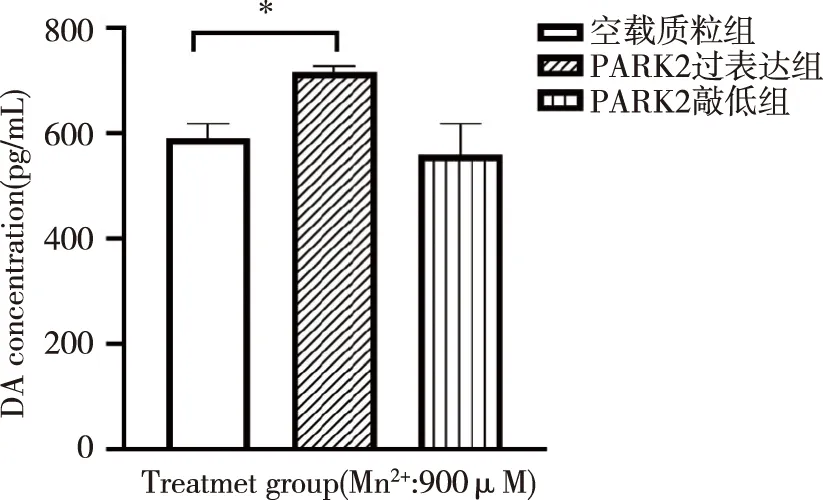
*:与空载质粒组比较,图7 不同处理组细胞培养基中DA水平
3 讨论
锰进入机体后,主要的作用靶点为大脑的多巴胺能神经细胞。研究组的前期研究[16-19]发现:锰暴露可引起人骨髓神经母细胞瘤细胞株(类多巴胺能神经细胞)PARK2 mRNA和Parkin蛋白的表达均降低,SOD活力下降,MDA含量增加,细胞氧化应激,机体清除氧自由基的能力下降,导致细胞凋亡增加,合成多巴胺减少;锰暴露大鼠全血、唾液、纹状体和皮质内的锰水平与PARK2/Parkin 表达呈负相关,高剂量染锰组多巴胺能神经元几乎消失;冶炼厂中的锰暴露可导致工人血液中PARK2的表达减少。本次研究发现通过构建PARK2基因过表达/敲低的细胞,继而改变Parkin蛋白的表达,从而影响细胞内线粒体自噬来清除受损的线粒体。这说明锰中毒的作用机制之一可能为通过降低PARK2基因的表达,阻碍细胞内线粒体自噬,从而导致细胞凋亡。
锰对线粒体有特殊的亲和力,可蓄积于富含线粒体的组织中,阻断能量合成,有研究表明锰中毒可导致线粒体代谢障碍[20]。细胞中线粒体自噬是细胞中清除失去功能的线粒体、维持细胞正常功能的重要途径,对维持细胞内稳态具有重要意义[12],一旦线粒体自噬受到影响,可导致细胞死亡。
PARK2基因表达产物为Parkin蛋白,Parkin蛋白是一种E3泛素蛋白连接酶,是泛素-蛋白酶体途径(UPP)中的重要组成成员,失去功能蛋白质在细胞中堆积可导致细胞的死亡,而UPP途径在清除细胞中失去功能的蛋白质过程中发挥重要作用[21]。PARK2基因突变可出现细胞内蛋白降解障碍,蛋白降解受阻并积聚可引起神经毒性,Kitada等发现该基因突变可导致常染色体隐性遗传性青少年型帕金森综合征,PARK2基因突变导致了多巴胺能神经元细胞中Parkin蛋白缺乏、UPP途径障碍、多巴胺能神经细胞死亡,导致青少年型帕金森综合征[22]。
自噬过程[23]分为 4 个阶段:(1)双层隔离膜形成;(2)自噬体形成;(3)自噬溶酶体形成;(4)自噬体溶酶体降解。Kimura等[24]研究发现自噬相关蛋白 LC3 是哺乳动物细胞中酵母自噬相关基因 8 (Autophagy-related gene,ATG8)的同源物,靶向定位于自噬体膜,分为 LC3Ⅰ和 LC3Ⅱ。当自噬发生时,细胞内形成自噬泡,LC3Ⅰ经泛素样加工修饰与自噬泡表面的磷脂酰乙醇胺(PE)形成 LC3Ⅱ,是自噬发生的标志性蛋白。Yamano等[25]研究发现OPTN和SQSTM1(P62)为哺乳动物的自噬受体,当线粒体自噬发生时,这些受体被招募到线粒体外膜上与 LC3-Ⅱ结合形成复合物,最终在溶酶体内降解,随着自噬作用的增加,自噬受体蛋白被不断消耗,LC3Ⅱ与自噬受体蛋白水平成反比,是细胞内自噬效应的标志蛋白之一。
在本研究中,使用SK-N-SH细胞作为染锰的研究细胞,该细胞与锰作用于人体中枢神经中的多巴胺能神经元细胞的功能类似。PARK2过表达细胞株和敲低细胞株用定量PCR和WB实验予以验证,结果证明试验所用的细胞株达到PARK2过表达和敲低的目的。
各组染锰后,PARK2过表达细胞株的多巴胺水平高于对照组和PARK2敲低组,说明细胞内Parkin水平的提高有利于细胞功能的正常发挥,对染锰细胞有保护作用;染锰后,随着自噬小体增加,OPTN和P62蛋白水平也降低,LC3Ⅱ/Ⅰ 升高,说明染锰对SK-N-SH细胞损害,由于线粒体自噬的增加而降低。证明在PARK2过表达的细胞中受损线粒体有较高水平的Parkin组成的UPP参与自噬,对细胞清除受损的线粒体,保持细胞内稳态有促进作用[26],从而降低了锰对细胞的毒性。
在PARK2敲低细胞组中,多巴胺水平与空载质粒组比较未见显著差异,可能原因是染锰后,锰引起细胞毒性时已经降低了Parkin蛋白的表达(和我们的前期研究结果一致),因此再对细胞PARK2基因敲低,其产生的效应对实验结果的影响不明显。虽然P62和OPTN蛋白表达量与空载质粒组比较增加具有显著性,自噬小体数量与空载质粒组比较,趋势有降低,但差异没有显著性。说明染锰产生的毒性和PARK2敲低同时作用,并未对自噬效应产生明显叠加效应。
本次实验发现过表达Parkin后OPTN蛋白表达降低,敲低Parkin后OPTN蛋白表达增高。推测在锰致细胞神经毒性时,OPTN与P62发挥相同作用:作为Parkin蛋白的自噬受体蛋白参与Parkin介导的线粒体自噬过程。我们的推测符合Wong等[27]的研究结果:通过高分辨活细胞成像发现OPTN蛋白被招募到Parkin泛素化的线粒体上,与LC3相互作用诱导自噬泡包裹受损线粒体形成自噬小体。
综合研究结果,我们可以推测:Parkin可能通过介导线粒体自噬效应减轻锰对神经细胞的毒性作用,并且OPTN蛋白作为Parkin蛋白的下游蛋白参与此过程。
