影响猪采食行为的肠道微生物种类鉴别
2023-02-03方绍明陈从英
姜 辉,杨 慧,方绍明,高 军,陈从英
(江西农业大学省部共建猪遗传改良与养殖技术国家重点实验室,南昌 330045)
采食是机体获取营养物质的重要行为模式[1],而饥饿与饱腹感的交替支配则是调控动物采食行为的内在机制[2]。采食行为主要包括两个阶段:食欲阶段(如觅食等)和反射行为完成阶段(如吞咽等)[3]。食欲控制可分为两种模式:一种与宿主能量稳态有关;另一种模式是基于细菌与宿主的交流。肠道微生物不仅可以通过调控宿主能量平衡来调节自身生长并维持其在肠道内的种群数量[2],还可以通过“脑-肠轴”对宿主行为进行干预。肠道微生物通过降解膳食纤维等难以被宿主消化吸收的物质,释放信号肽等代谢产物并作用于中枢神经系统,从而实现对宿主食欲的调节[4]。因此,肠道微生物在大脑与肠道之间的双向交流中发挥重要作用[2, 4-5],影响宿主的采食行为。随着相关研究的不断深入,将为通过肠道微生物调控宿主能量摄入与平衡提供新策略。近年来研究发现,肠道微生物在宿主采食行为的调节中起重要作用。Li等[6]通过比较不同分组的奶牛群体肠道微生物组成,发现高采食组的拟杆菌门(Bacteroidetes)与厚壁菌门(Firmicutes)的比例显著低于中低采食组。当肠道微生物代谢途径发生改变,代谢产物(信号分子)直接或间接(前体物质)介导中枢神经系统,进而改变宿主食欲[7-10]。Breton等[11]发现,大肠杆菌经营养物质诱导后产生的细菌蛋白质可通过激活大脑的饱腹感影响宿主食欲。Yang等[12]研究表明,以普氏菌属(Prevotella)为肠型的杜洛克猪平均日采食量(average daily feed intake, ADFI)要高于密螺旋体(Treponema)肠型。研究发现,肠道部分微生物产生的短链脂肪酸(short-chain fatty acids, SCFAs)不仅能为肠上皮细胞提供能量,维持稳态[13],而且可以刺激肠道内分泌L细胞释放饱感激素(如PYY、GLP-1等)[14],最终降低宿主食欲。此外,革兰阴性菌和革兰阳性菌都可以产生大量的细胞间信号分子——吲哚[15],吲哚可以通过调节胃肠道激素的分泌而影响宿主食欲和代谢过程[16]。然而,猪肠道中哪些微生物种类能调节宿主采食行为目前还知之甚少,有待深入研究。
1 材料与方法
1.1 试验猪饲养与表型测定
本研究选取210头未经药物治疗的健康商业杜洛克猪作为研究材料,其中包含72头母猪和138头公猪。试验猪群体采用相同的饲养和管理方式并在28日龄断奶。当仔猪体重达到30 kg后转移到配备奥斯本自动投喂器(美国)的育肥舍进行饲养并饲喂相同的商业配方饲料。饲料以玉米、豆粕、大豆油、赖氨酸和磷酸氢钙为主,其中营养水平为:粗蛋白15%、粗脂肪1.5%、粗纤维5%、粗灰分6%、赖氨酸0.8%、钙0.9%、磷0.5%、盐0.3%。饲料和水可自由获取。使用奥斯本自动投喂器记录测定期间试验猪(30~100 kg(90~170日龄))的日采食量、日增重、日采食时间、日采食次数等表型,并进一步计算平均日采食时间(average daily eating time,ADET)、平均日采食次数(average daily eating visits,ADEV)、平均日采食量(average daily feed intake,ADFI),剩余采食量(residual feed intake,RFI)[17],平均日增重(average daily gain,ADG)等表型值。采用B超仪测定试验猪的背膘厚(backfat,BF)。
1.2 粪便样品的采集与16S rRNA基因测序
分两个批次(2017年3月20日与4月20日)采集140日龄左右试验猪粪便样本。从肛门处采集新鲜粪便样品放入1.5 mL无菌离心管中,随后迅速浸入液氮并于-80 ℃冰箱保存。使用QIAamp Fast DNA Stool Mini Kit(Qiagen, Germany)进行粪便DNA提取[18]。用Nanodrop 1000超微量分光光度计测定DNA样品的浓度和纯度,并使用0.8%琼脂凝胶电泳进行DNA质量检测。按照1∶10的比例将DNA样品进行稀释,取1 μL稀释后的DNA样品用于PCR扩增。本研究选用细菌通用引物338F(5′-ACTCCTACGGGAGGCAGCA-3′)和 806R(5′-GGACTACHVGGGTWTCTAAT-3′)对16S rRNA基因的V3-V4区进行扩增[19-20]。根据标准化操作流程利用纯化后的PCR扩增产物构建双端测序文库,使用MiSeq测序平台(Illumina,USA)进行测序。
1.3 16S rRNA基因测序数据处理
使用Trimmomatic(V.0.39)软件对原始测序数据中的引物和标签序列进行过滤[21],采用FLASH(v.1.2.11)合并双端配对的clean reads得到tags[22],利用USEARCH(v.7.0.1090)软件去除嵌合序列[23]。为消除不同测序深度对分析结果的影响,将每个样本得到的拼接序列抽平至16 000个tags。用VSEARCH软件(v.2.8.1)依据97%的序列相似度将获得的tags聚类成可操作分类单元(operational taxonomic units,OTUs)[24]。利用RDP classifer(v2.2)对每条基因序列进行物种注释[25],并使用MOTHUR(v1.31.2)软件计算肠道微生物的alpha多样性[26]。基于16S rRNA基因测序数据,通过PICRUSt(v1.0.0)软件对肠道微生物潜在功能进行预测,并依据16S rRNA基因序列,计算Kyoto Encyclopedia of Genes and Genomes(KEGG)通路的相对丰度[27]。
1.4 OTU共丰度组(co-abundance groups,CAGs)分析
将相对丰度高于1×10-4,至少在20%个体中出现的共计748个OTUs用于后续分析。首先,基于SparCC算法[28]计算OTU之间的相关性并将其转换为相关性距离矩阵。利用R包中hclust功能(ward.D2算法)[29]将距离矩阵进行聚类,采用多因素方差分析(PerMANOVA)对CAGs彼此间的差异性 (P<0.05) 进行置换检验[30]。最后,使用Cytoscape(version 3.7.1)对权重值大于0.5的OTUs进行网络可视化展示[31]。使用R包中的corr.test函数计算每个CAG与表型ADFI值之间的Spearman相关性系数[17]。
1.5 使用Two-part模型分析肠道菌群与采食行为表型值的相关性
使用R(4.0.0)软件psych包中的corr.test函数基于Spearman相关性检验分析表型之间的相关性[32]。为探索肠道微生物与宿主采食行为之间的相关性,采用Two-part模型对过滤后的OTUs与各表型值进行关联分析[33]。该模型的优势在于同时考虑了二元模型与数量模型,克服了微生物数据非正态分布的难题。二元模型用于检测肠道微生物存在与否对表型值的影响,表达公式为:y=β1b+e,y为经过相应性别、批次校正后的表型值,β1表示二元模型的估计效应,b代表二元特征(0/1),e是残差;数量模型则解释了微生物的丰度对表型值的影响,表达公式为y=β2q+e,y为经过相应性别、批次校正后的表型值,β2表示数量模型的估计效应,q代表数量特征(相对丰度),e是残差。随后使用未加权Z方法整合二元模型与数量模型结果执行荟萃分析并获得metaP值。最终P值为二元分析、数量分析以及荟萃分析所得P值的最小值,Z值是基于Z分布计算而来。该模型还采用了1 000次排列检验的方法将错误值(false discovery rate,FDR)控制在0.05水平。
1.6 采用LEfSe方法鉴定采食行为表型极端个体间的差异OTU
分别挑选ADFI值最高和最低各10个个体,使用在线(https://huttenhower.sph.harvard.edu/galaxy/)线性判别分析效应大小(linear discriminant analysis effect size,LEfSe)软件分析两组极端个体间差异的OTU,将linear discriminant analysis(LDA)差异显著性阈值设定为2.5。
1.7 猪采食行为相关的潜在肠道菌群功能通路与表型值显著关联的OTUs之间的相关性分析
采用Spearman相关性分析鉴定与ADFI表型值显著相关的KEGG代谢通路,并将显著关联的功能通路与上述Two-part模型分析发现的与ADFI表型值显著相关的25个OTUs进行Spearman关联分析。使用R包pheatmap(version 1.0.12)绘制相关性热图[34]。
2 结 果
2.1 试验猪群体采食行为各表型间相关性
2.1.1 试验猪群体采食行为表型值分布概况 本研究以210头商业杜洛克猪为对象测定的ADET值(min·d-1)和ADFI值(kg·d-1)均符合正态分布,平均值用“均值±标准差”表示,分别为3 891.87±566.79和2.22±0.20(图1A、1C),而ADEV值(次·d-1)不符合正态分布,平均值为6.22±2.16(图1B)。

A. ADET表型分布;B. ADEV表型分布;C. ADFI表型分布;D. 纵坐标表示表型间的相关性系数,红色为正相关,蓝色为负相关A. Phenotype distribution of ADET; B. Phenotype distribution of ADEV; C. Phenotype distribution of ADFI; D. The Y-axis represents the correlation coefficients between phenotypes. Red is positive correlation, blue is negative correlation图1 210头试验猪采食行为相关表型值的分布及表型间的Spearman相关性分析Fig.1 Distribution of phenotypic values related to feeding behaviors in 210 experimental pigs and the correlation between phenotypes by spearman correlation analysis
2.1.2 采食行为各表型间及其与平均日增重、剩余采食量、背膘厚间的相关性分析结果 试验猪群体各采食行为表型间(ADET、ADEV、ADFI)及其与ADG、RFI、BF之间的斯皮尔曼相关性分析结果(图1D)表明,ADET分别与ADEV(r=0.41,P<0.05)、RFI(r=0.32,P<0.05)呈显著正相关,ADFI分别与ADG(r=0.63,P<0.05)、RFI(r=0.56,P<0.05)呈显著正相关,另外,RFI与BF显著正相关(r=0.16,P<0.05),然而ADFI与ADET、ADEV以及BF之间无显著相关性。
2.2 不同分类水平上试验猪的肠道微生物组成概况
2.2.1 16S rRNA基因测序结果 将收集到的210份试验猪粪便微生物DNA样品全部进行16S rRNA基因测序。获得的原始测序数据上传至中国国家基因库(China National Gene Bank),序列号为CNP0000828。质控后,每个样品平均可得到34 411个高质量tags,按照97%的序列相似性聚类后,平均每个样本可以获得874个OTUs。OTU注释结果显示,检测到的肠道微生物共涉及19个门、70个科和94个属。α多样性结果如图2所示(数据表示为“均值±标准差”),Ace指数为986.393±84.212,Chao指数为1002.035±85.918,Shannon指数为5.131±0.289,Simpson指数为0.017±0.009(表1)。
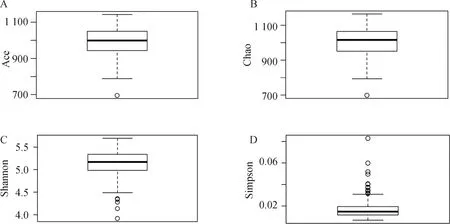
图2 试验猪群体肠道微生物α多样性Fig.2 Alpha-diversity of gut microbiota in experimental pig population
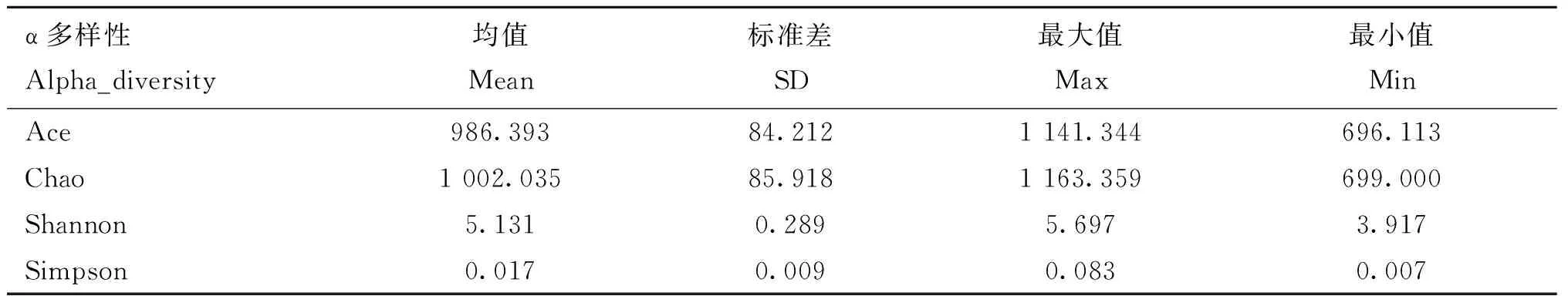
表1 试验猪群体肠道微生物α-多样性指数Table1 Alpha-diversity index of gut microbiota in experimental pig population
2.2.2 不同分类水平下肠道微生物组成概况 试验猪肠道微生物中丰度最高的门依次是厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)和变形菌门(Proteobacteria)。在科水平上,丰度最高的科分别是瘤胃球菌科(Ruminococcaceae)、普雷沃氏菌科(Prevotellaceae)和毛螺菌科(Lachnospiraceae)。而在属水平上,丰度最高的是普氏菌属(Prevotella)、颤螺旋菌属(Oscillospira)和乳酸杆菌属(Lactobacillus)(图3)。
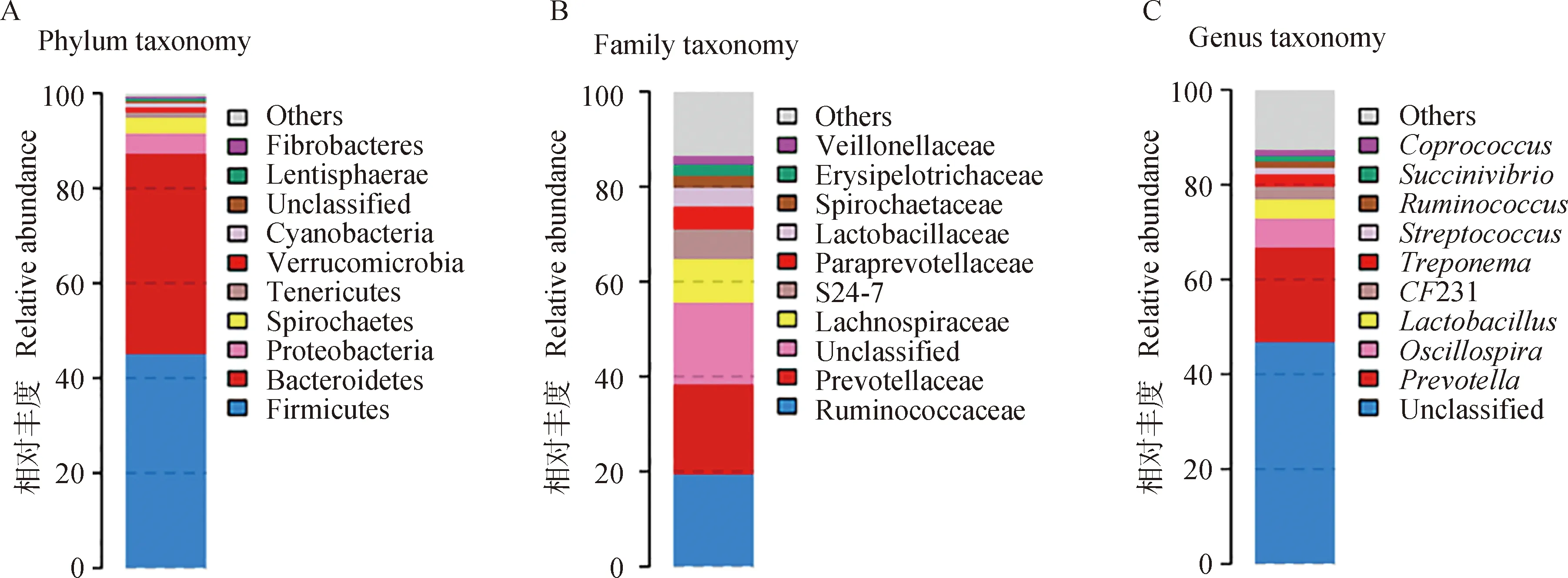
图3 基于16S rRNA测序的门水平(A)、科水平(B)和属水平(C)上肠道微生物的组成Fig.3 The composition of gut microbiota at phylum level(A), family level(B) and genus level(C) base on 16S rRNA gene sequencing
2.3 CAGs分析结果
基于SparCC方法将过滤后的748个OTUs聚类成9个CAGs(图4A)。每个CAG内的OTU彼此之间均为正相关,而不同的CAG中的OTU彼此之间则呈负相关。在9个CAG中,发现CAG2、CAG9与采食行为表型中的ADFI显著相关(P<0.05)。
Spearman相关性分析结果表明CAG2与ADFI值呈显著负相关(P<0.05,图4B),该CAG中包含118个OTUs,这些OTUs主要注释到拟杆菌目(Bacteroidales)、瘤胃球菌科和颤螺菌属等(图4A)。CAG9与表型呈显著正相关(P<0.05,图4B),该CAG中包含70个OTUs,其中有5个OTUs可以注释到物种水平,分别是柔嫩梭菌(Faecalibacteriumprausnitzii)、两形真杆菌(Eubacteriumbiforme)和Asteroleplasmaanaerobium(图4A)。然而并未发现与其他采食行为表型相关的CAG。
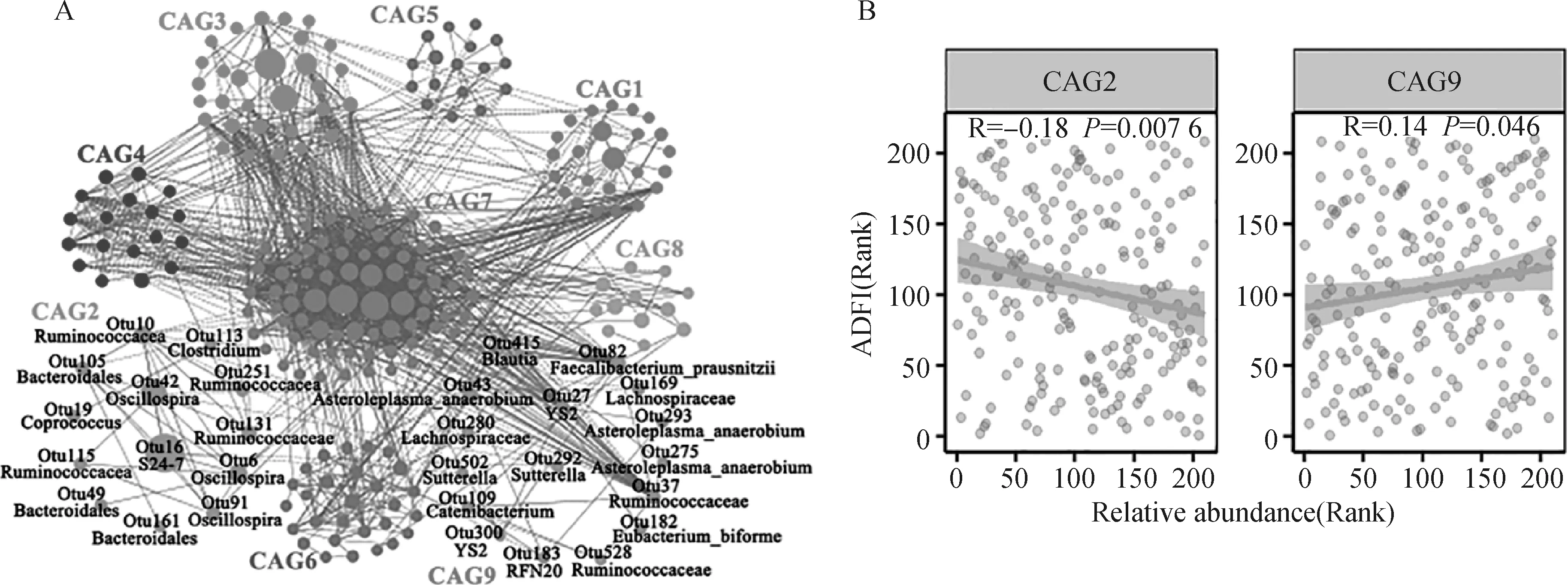
图4 CAGs分析结果可视化展示(权重值>0.5)Fig.4 Visual display of CAGs analysis results (weight>0.5)
2.4 与猪采食行为相关的肠道微生物种类
2.4.1 协变量对表型值的影响 Wilcoxon秩检验分析结果表明,在本研究群体中,性别、批次对ADET值均具有显著影响(P<0.05),只有批次会对ADEV值产生显著性影响(P<0.05),而性别、批次对ADFI值均无显著性影响(P>0.05),无需进行任何校正(图5)。针对ADET值,将性别、批次作为因子变量纳入线性混合模型后,经过999次置换检验,发现两者均会对表型产生显著性影响(P值分别为0.013和0.018)。同样地,对于ADEV值,只有批次会对表型值产生显著性影响(P=0.001),而性别则无显著性影响(P=0.069)。而对于ADFI值,性别、批次均不会对表型值产生显著性影响(P值分别为0.337和0.768)。与Wilcoxon秩检验分析结果相一致,验证了数据分析的可靠性。
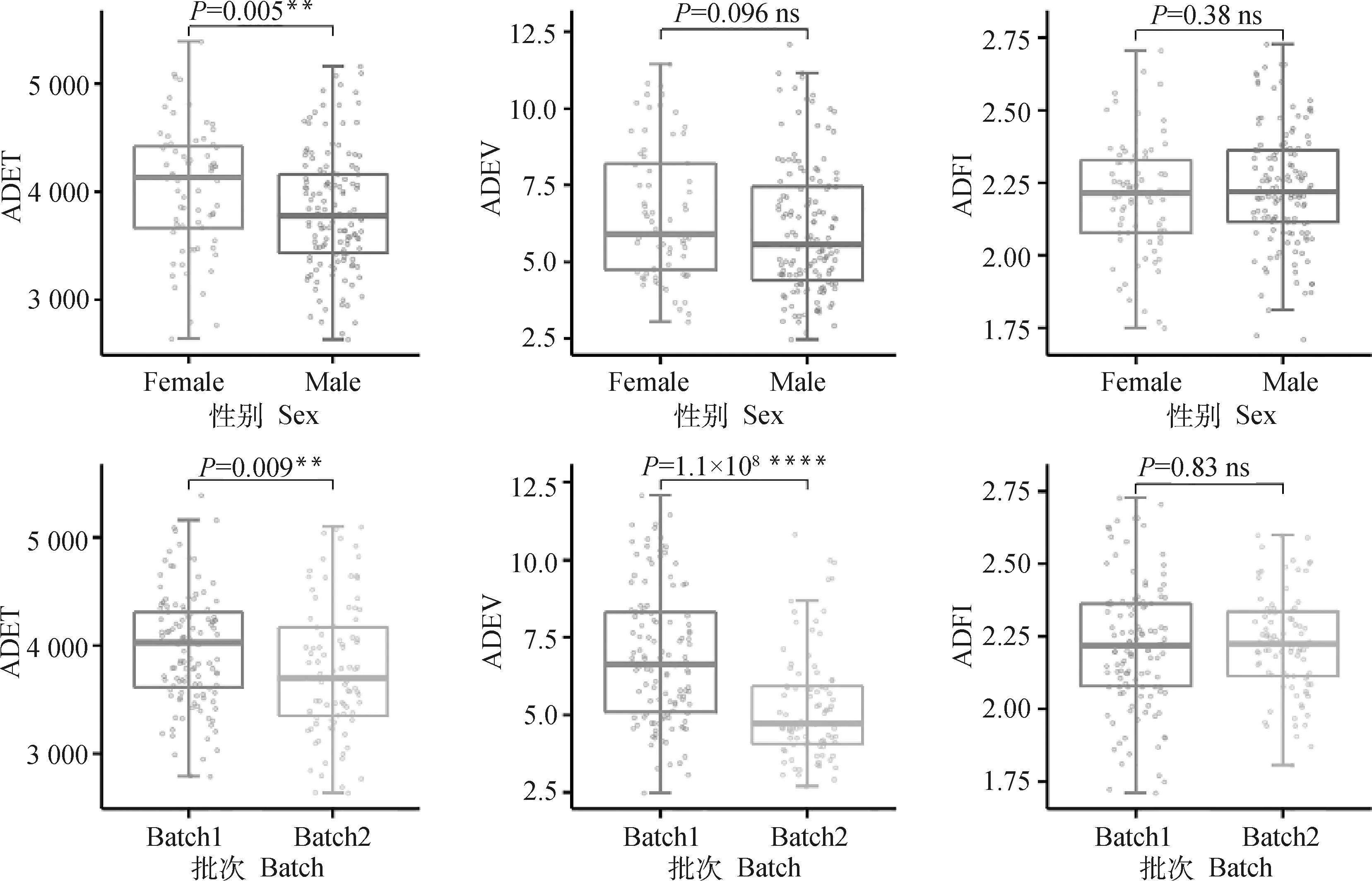
**、****表示差异显著(P<0.05);ns表示差异不显著**,**** mean significant different (P<0.05); ns means not significant different图5 基于Wilcoxon秩检验分析性别、批次对表型值的影响Fig.5 Effects of sex and batch on phenotypic values based on Wilcoxon rank test
2.4.2 与ADET相关的肠道菌群种类 采用Two-Part模型分析试验猪各表型值与肠道微生物种类的相关性,FDR<0.05时,未鉴定到任何与采食行为显著相关的OTU,但在P<0.01时,一共有14个OTUs与ADET呈现相关性,其中有7个OTUs与ADET呈正相关,剩余的7个OTUs则与ADET呈负相关(P<0.01,图6A、表2)。这14个OTUs中注释到门水平1个,目水平有3个,科水平有8个,属水平有2个。

表2 与ADET表型值关联的OTUs(P<0.01)Table 2 OTUs associated with the phenotypic values of ADET (P<0.01)
2.4.3 与ADEV相关的肠道菌群种类 从OTU与ADEV相关性分析结果发现:一共有21个OTUs与ADEV呈现出相关性趋势,其中有13个OTUs与ADEV正相关,另8个OTUs与ADEV呈负相关(P<0.01,图6B,表3)。在这21个OTUs中可以注释到目水平的有6个,科水平的有9个,属水平的有5个,剩下的1个OTU被注释为生黄瘤胃球菌(Ruminococcusflavefaciens)。

表3 与ADEV表型值关联的OTUs(P<0.01)Table 3 OTUs associated with the phenotypic values of ADEV (P<0.01)
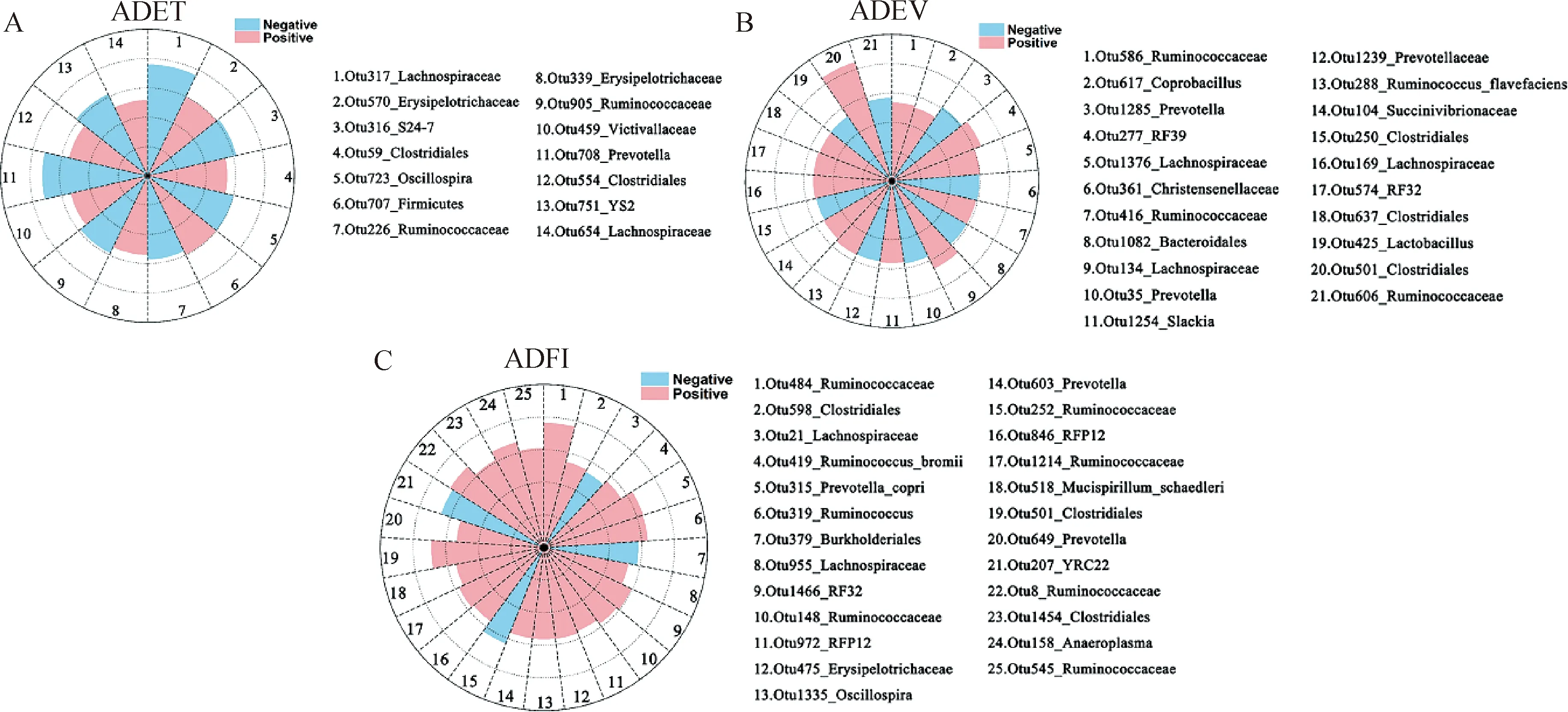
扇形面积表示Z值绝对值,红色为正值,蓝色为负值The sector area represents the absolute value of the Z-score, with positive values in red and negative values in blue图6 与采食行为具有相关性趋势的OTUsFig.6 OTUs showed trends of correlation with feeding behaviors
2.4.4 与ADFI相关的肠道菌群种类 共检测到25个OTUs与ADFI具有相关性趋势,其中21个OTUs与ADFI正相关,剩下的4个OTUs与ADFI呈负相关(P<0.01,图6C,表4)。在这21个OTUs中,有5个被注释到瘤胃菌科,有3个注释到梭菌目(Clostridiales),有2个注释到RFP12, 2个注释到普氏菌属,其余9个分别被注释到布氏瘤胃球菌(Ruminococcusbromii)、Prevotellacopri、Mucispirillumschaedleri、瘤胃球菌属(Ruminococcus)、颤螺菌属、厌氧支原体属(Anaeroplasma)、RF32、毛螺菌科和韦荣球菌科(Erysipelotrichaceae)。与ADFI呈负相关的4个OTUs分别注释到毛螺菌科、伯克氏菌目(Burkholderiales)、瘤胃菌科和YRC22。

表4 与ADFI表型值相关的OTUs(P<0.01)Table 4 OTUs associated with the phenotypic values of ADFI (P<0.01)
2.5 ADFI表型极端个体间显著差异的OTUs
选择ADFI表型极端个体,采用LEfSe方法鉴定High_ADFI与Low_ADFI组间具有显著差异的OTUs。将LDA显著性阈值设定为2.5时,High_ADFI组共富集10个OTUs,其中2个注释到普氏菌属,2个注释到瘤胃球菌科,2个注释到拟杆菌目,其余4个分别被注释到毛螺菌科、梭菌目、CF231和S24-7。Low_ADFI组共富集14个OTUs,3个被注释到克里斯滕森菌科(Christensenellaceae),其余11个OTUs分别注释到Mogibacteriaceae、伯克氏菌目、R4-45B、普氏菌属、YRC22、拟杆菌目、RFN20、密螺旋体科、Sphaerochaeta、瘤胃球菌属和瘤胃球菌科(图7)。
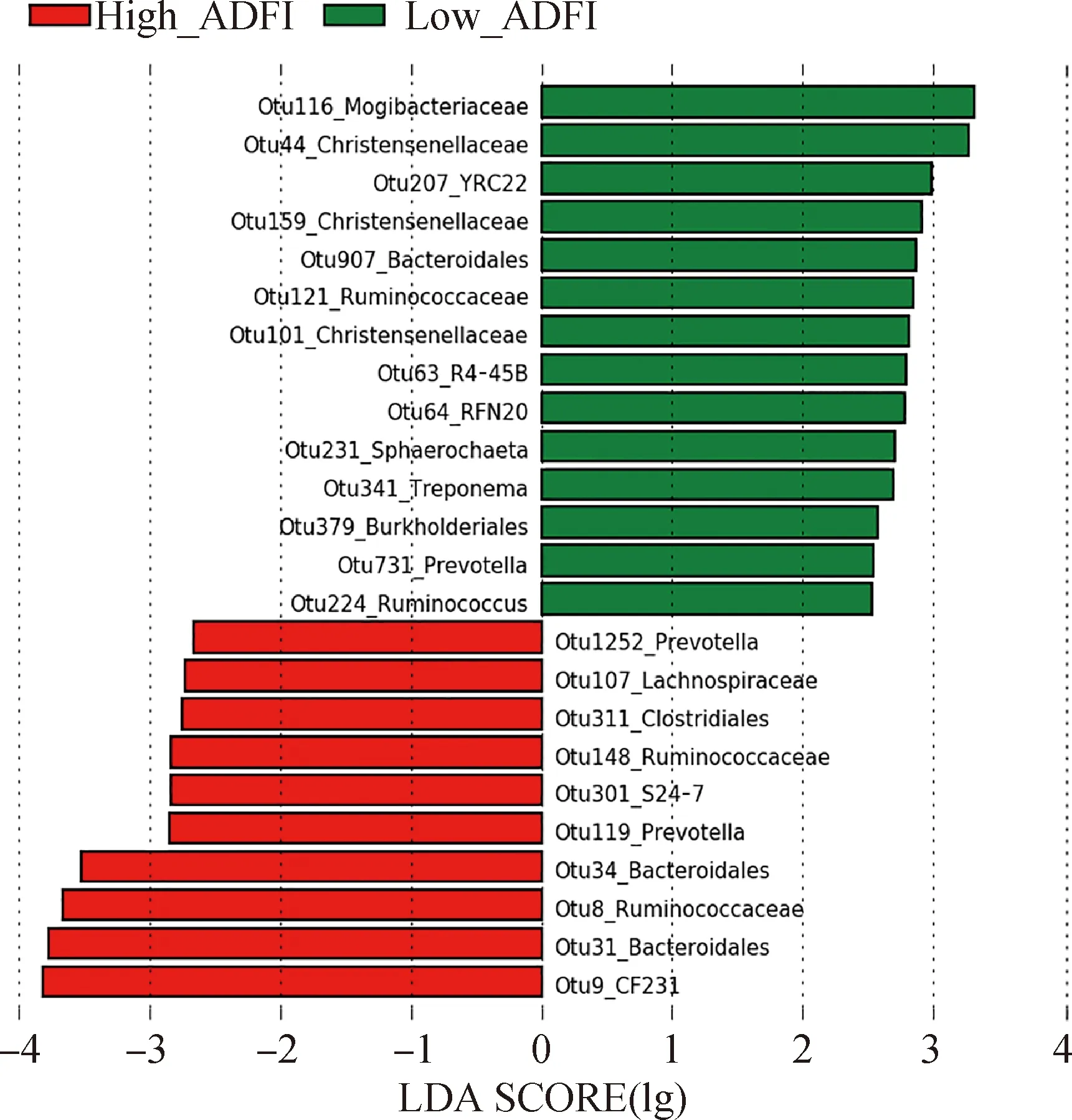
图7 高低ADFI极端个体中显著差异的OTUs(LDA>2.5)Fig.7 OTUs showing significantly different abundances between high and low ADFI pigs (LDA score>2.5)
2.6 与采食行为相关的肠道微生物功能
2.6.1 与ADFI值显著关联的肠道微生物功能通路 通过Spearman相关性分析,当P<0.05时,鉴定到鞘脂代谢(Sphingolipid metabolism),糖胺聚糖降解(Glycosaminoglycan degradation),其他聚糖降解(Other glycan degradation)等6个功能通路与ADFI表型值呈显著负相关。而胰岛素信号通路(Insulin signaling pathway)和脂质代谢(Lipid metabolism)等14个功能通路则与ADFI值呈显著正相关(表5)。

表5 ADFI表型值与微生物功能通路之间的相关性分析结果Table 5 The correlation between ADFI phenotypic values and predicted function pathways
2.6.2 ADFI相关的微生物功能通路与ADFI相关的OTUs之间的关联性 如图8所示,与试验猪ADFI呈负相关的4个OTUs(毛螺菌科、伯克氏菌目、瘤胃菌科和YRC22)(图6C)与溶酶体(Lysosome)和/或鞘脂代谢功能通路显著正相关(P<0.05),而这些功能通路均与ADFI值负相关(P<0.05,表5)。其余21个与ADFI值正相关的OTUs(图6C),与胰岛素信号通路等促进试验猪ADFI的功能通路整体呈显著正相关(P<0.05,表5),而与那些抑制猪ADFI的功能通路呈显著负相关(P<0.05,表5)。
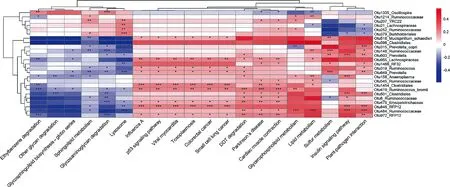
*、**、***表示显著相关(P<0.05)*,**,*** mean significantly correlated (P<0.05)图8 与ADFI值相关的代谢通路和OTUs之间的相关性分析Fig.8 The correlations between ADFI-associated predicted functional pathways and ADFI-associated OTUs
3 讨 论
本研究相关性分析表明,ADFI分别与ADG、RFI存在显著的正相关性,表明猪采食量的增加可以促进其生长发育,但是有降低饲料效率的风险。ADET和ADEV之间也呈显著正相关关系,但二者均与ADFI之间不存在显著关联,这种情况最合理的解释可能是猪活动过程中误触了自动饲喂器但并没有真正进食,使得猪真正的采食时间和采食次数记录不够准确。
本试验中,猪的肠道微生物主要是由厚壁菌门、拟杆菌门和变形杆菌门等构成,与之前的研究结果类似[35-36]。Spring等[37]研究结果表明,瘤胃球菌科、普雷沃氏菌科和毛螺菌科是各饲粮组猪粪便中的优势菌群。多项研究发现,普氏菌属、颤螺菌属以及乳酸杆菌属通常是猪的核心菌属[38-39],说明了本研究中16S rRNA测序结果的可靠性。
CAGs分析中,鉴定到与ADFI负相关的微生物主要是瘤胃球菌科、拟杆菌目以及颤螺菌属。多项研究发现,拟杆菌目具有碳水化合物降解活性,能够发酵多种植物多糖,从而产生乙酸等SCFAs[40-41]。同样地,难以被内源性酶消化的纤维素等物质被部分瘤胃球菌科细菌酵解时可产生SCFAs,而SCFAs不仅有助于机体维持稳态[42],还能够刺激肠内分泌L细胞释放饱感激素PYY(peptide YY)和GLP-1(glucagon-like peptide 1)[14],从而抑制宿主采食。另外,一些颤螺菌属可能是丁酸产生菌[43-44],能够在抑制食欲方面发挥重要作用[45]。分析发现,与ADFI正相关的CAG9中有4个OTUs可以注释到物种水平,除之前研究发现的柔嫩梭菌外[12],本研究还鉴定到韦荣球菌科中的两形真杆菌和Asteroleplasmaanaerobium具有增加宿主采食量的潜能。韦荣球菌科往往与肥胖相关[46],有趣的是,表型相关性分析结果表明ADFI与BF具有正相关趋势(r=0.1,P=0.19),说明韦荣球菌科在增加采食量的同时会导致脂肪沉积,与本文相关研究结果相吻合。
Two-part模型与CAGs分析结果基本一致,说明本研究分析结果的可靠性。除之前发现的YRC22具有抑制宿主食欲的潜能外[12],本研究还发现,伯克氏菌目可通过产SCFAs[47]参与神经体液调节通路,最终抑制宿主进食。与此高度吻合的是,本试验在挑选ADFI极端值而进行的LEfSe差异分析中同样鉴定到这两种肠道微生物在Low_ADFI组显著富集。而毛螺菌科除产生丁酸外还可以产生乳酸[48],本研究也发现乳酸菌与ADEV呈负相关,且乳酸菌一直以来通过产生乳酸而促进肠道发育和代谢并充当益生菌的角色[49]。有研究表明,宿主体内乳酸的产生能够激活其饱腹感通路从而宿主减少对食物的摄入[50]。而与食欲呈正相关趋势的OTUs主要注释为普氏菌属(尤其是Prevotellacopri)、厌氧支原体属、韦荣球菌科以及部分瘤胃菌科和毛螺菌科。另外,Myer等[51]研究发现,厌氧支原体属和粪杆菌属(Faecalibacterium)在ADGHigh-ADFIHigh类群中数量最多,在本研究中亦有相似发现,且ADFI与ADG显著正相关。研究发现,普氏菌与富含丝氨酸、半胱氨酸等生物合成的KOs呈正相关,且在高ADFI组具有更高的丰度[12],不仅如此,普氏菌属与一种开胃激素相关联,与其在促进宿主食欲方面的功能至关重要[52]。值得一提的是,本研究中LEfSe组间差异分析结果显示,Low_ADFI组显著富集的14个OTUs中有3个被注释为克里斯滕森菌科。研究表明,克里斯滕森菌科属于革兰阳性菌,能够产生SCFAs[53]。不仅如此,Goodrich等[54]研究发现,克里斯滕森菌科在低身体质量指数(body mass index,BMI)个体中富集。这些研究结果刚好与本研究中ADFI与ADG显著正相关的发现相一致。
最后,本研究通过对肠道微生物进行功能预测分析发现,胰岛素信号通路与ADFI值显著正相关。之前的研究表明,胰岛素水平的提高会增加饥饿感,增强机体对甜味的感知,增加食物的摄入量[55]。瘦素是一种主要由脂肪细胞合成和分泌的蛋白质,可以调节食欲,并在脂肪酸氧化等脂质代谢过程中发挥重要作用[56]。这在一定程度上可以解释本研究中脂质代谢与ADFI值之间的显著生物学相关性(P<0.05)。一磷酸鞘氨醇(sphingosine-1-phosphate,S1P)是鞘脂代谢(Sphingolipid metabolism)中间产物,S1P能够作用于后肠特异性神经肽样受体dNepYr的上游,并通过dNepYr信号通路抑制食欲[57]。有趣的是,本研究中同样发现鞘脂代谢与ADFI值显著负相关。不仅如此,本研究还发现分别与ADFI值关联的代谢功能和肠道微生物之间也呈现显著相关性,即与ADFI值相关性方向一致的代谢功能和OTU之间呈显著正相关,否则呈显著负相关。这说明肠道微生物对宿主表型的影响与其功能相适应。
4 结 论
本研究表明,一些细菌(如伯克氏菌目、瘤胃球菌科、克里斯滕森菌科、毛螺菌科等)因产生SCFAs或乳酸可能在抑制猪采食量方面发挥重要作用,而柔嫩梭菌、Asteroleplasmaanaerobium和普氏菌属(尤其是Prevotellacopri)则可以增加宿主采食量,可能是控制宿主食欲的关键微生物。但是本研究存在一定的局限性,受限于16S rRNA测序的分类水平,很多OTUs只能注释到属水平甚至科水平,因此无法明确微生物物种对采食行为的影响及其作用机制,今后仍需结合宏基因组和代谢组进一步深入地研究并验证潜在的机制假说。
