基于全基因组填充重测序关联分析鉴别影响苏山猪初生体尺与乳头数性状的遗传位点
2023-02-03张昌政李德森方晓敏赵为民任守文董焕声周李生
张昌政,李德森,黄 敏,方晓敏,赵为民,任守文,董焕声 任 军 周李生*
(1.青岛农业大学动物科技学院,青岛 266109; 2.华南农业大学动物科学学院,广州 510642;3.江苏省农业科学院畜牧研究所,南京 210014)
猪的初生体尺和乳头数性状是衡量猪生长和繁殖性能的重要指标,直接影响养猪业的经济效益。已有研究表明,初生体尺作为衡量仔猪生存能力和生长发育的可靠预测指标,与后期的生长速度和成活率呈显著正相关[1-3];初生重较高的仔猪在达到屠宰体重后,具有更好的胴体品质[4]和更优秀的体尺特征[5]。有效乳头数是评价母猪哺乳能力的基础[6],当能繁母猪的有效乳头数无法满足后代的哺乳需求,仔猪死亡率将显著增加,从而造成较大的经济损失[7-8]。同时,乳头数与产仔数存在一定程度的表型相关和遗传相关[9-10],核心母猪群体的平均有效乳头数与产仔数存在较为一致的趋势[11]。因此,鉴别影响仔猪的初生体尺和乳头数性状的分子标记有助于猪生长和繁殖性能的遗传改良。
苏山猪是江苏省农业科学院畜牧研究所团队利用太湖猪、丹麦长白猪和英国约克夏猪杂交育成的优质瘦肉猪新品种。本研究采用全基因组重测序结合基因型填充策略,在苏山猪群体中开展Fst与GWAS分析,挖掘与初生体尺和乳头数性状显著关联的位点与候选基因,为探究影响猪生长发育和乳头数形成的遗传机制提供借鉴,且为苏山猪核心种群的进一步选育提高奠定理论基础。
1 材料与方法
1.1 样本采集及表型记录
试验群体为269头苏山猪初生仔猪,均饲养于江苏省农业科学院六合基地实验猪场。仔猪在出生后24 h内测定并记录体重(body weight, BW)、体长(body length, BL)、胸围(chest circumference, ChC)、管围(cannon circumference, CaC)、左乳头数(number of teats on the left side, LTN)、右乳头数(number of teats on the right side, RTN)、总乳头数(total teat number, TTN)表型数据,并采集耳组织样品保存于含75%乙醇的2 mL离心管中。
体尺的测量方法:1)体长:在猪自然站立状态下,使用软尺自两耳连线的额顶中心起,沿背线至尾根紧贴体表量取;2)胸围:在猪自然站立状态下,切于肩胛骨后缘用软尺测量胸部垂直周径,软尺自然贴近体表为宜;3)管围:在猪自然站立状态下,使用软尺测量左前肢管部最细处的周径,测量时软尺紧贴体表。
1.2 DNA提取、基因分型和质量控制
使用常规酚氯仿法从猪耳组织样本中提取基因组DNA,使用Affymetrix Porcine SNP 55 K Array进行基因分型,并用Plink v1.9软件对所得SNP数据进行质量控制,标准:剔除SNPs检出率(call rate)小于90%,次等位基因频率(minor allele frequency, MAF)小于0.01,最后得到39 032个SNPs用于后续分析。
1.3 全基因组重测序分析
在269头苏山猪中挑选20个能够代表群体全部血缘的个体,采用Illumina NovaSeq platforms 的150 bp paired-end libraries,进行全基因组重测序(平均测序深度为20×)。此外,在NCBI(https://www.ncbi.nlm.nih.gov/)上下载了367头猪重测序数据,其中包括184头中国地方猪,13头中国野猪,142头欧洲商品猪,21头欧洲野猪和7头外群猪种。针对测序和下载的共387个个体的原始数据(raw data)进行质控,得到能满足数据分析要求的有效数据(clean data)。
1.4 变异位点检测
1.5 基因型填充
将269头苏山猪的55K SNP芯片数据作为目标数据集,387个重测序个体的SNP数据集作为参考数据集,利用Beagle软件进行基因型填充。随后利用等位基因准确性(allelic correct rate, CR)和基因型相关系数(correlation)对20头同时具有芯片数据和重测序数据的苏山猪进行基因型填充准确度的估计。最后综合基因型准确度和SNP的分布密度,保留call rate>0.9、MAF>0.01和correlation>0.8的SNPs用于后续分析。
1.6 选择信号分析
按照每个性状表型值的前10%和后10%分别分成两个亚群,基于填充后的SNP数据集,利用VCFtools v0.1.17软件,窗口大小为50 kb,以20 kb步长对每个SNPs进行滑窗计算Fst值。基于Fst的结果,使用R语言的Cmplot程序包绘制曼哈顿图,Fst阈值为按大小排序Fst值的前1%,高于阈值的点视为显著分化的区域。
1.7 全基因组关联分析
基于基因型填充后的SNP数据集,利用GEMMA软件对苏山猪的7个性状进行GWAS,为了校正由于亲缘关系、性别以及胎次等其他因素对关联分析造成的影响,本研究使用混合线性模型(mixed linear model, MLM)进行分析,模型如下:
y=Wα+Xβ+u+ε
y为表型向量;W为协变量(固定效应)的关联矩阵,将体重、性别和胎次作为初生体尺性状的协变量;α为包括截距在内的相应系数的向量;X为SNP矩阵;β为SNP的效应;u为一个m×1的随机效应向量,其中u~MVNm(0,λτ-1K),τ-1为残差的方差,λ为τ-1和ε的比值,K为n×n亲属关系矩阵;ε为随机残差的向量,ε~MVNn(0,τ-1In),In为单位矩阵;MVNn表示n维多元正态分布。
Bonferroni方法常被用来界定显著性阈值:0.05/N,N为研究SNPs的总数。考虑到Bonferroni校正对于本研究过于严格,则根据前人研究自定义P=1×10-5[12]为显著阈值线。使用R语言的Cmplot程序包对GWAS结果进行可视化。
1.8 基因功能富集分析
基于GWAS和Fst的结果,筛选出共有的显著SNPs,以这些SNPs为中心,利用R语言的GALLO程序包,参考基因组为11.1版本的猪基因组(Sus scrofa 11.1),在其上、下游500 kb范围内搜寻候选基因。为更好的了解基因的功能,精准地鉴别出与目标性状相关的候选基因,本研究利用Metascape(http://metascape.org)数据库对搜寻到的基因进行GO(gene ontology)分析和KEGG(kyoto encyclopedia of genes and genomes)通路分析,搜寻与目标性状相关的通路。利用在线富集分析可视化网站(http://www.bioinformatics.com.cn)对显著富集的基因绘制柱状图和气泡图。
2 结 果
2.1 表型描述统计
本研究统计269头苏山猪的7种表型的平均值、标准差、最大值、最小值和变异系数,结果如表1所示。结果显示,体重、体长、胸围、管围、左乳头数、右乳头数和总乳头数性状的变异系数为26.87%、8.97%、10.31%、9.54%、12.28%、12.69%和11.63%,其中体重的变异系数最大,高达26.87%,表明体重数据中存在较大的变异。基于标准差可以看出,右乳头数的变异程度大于左乳头数。
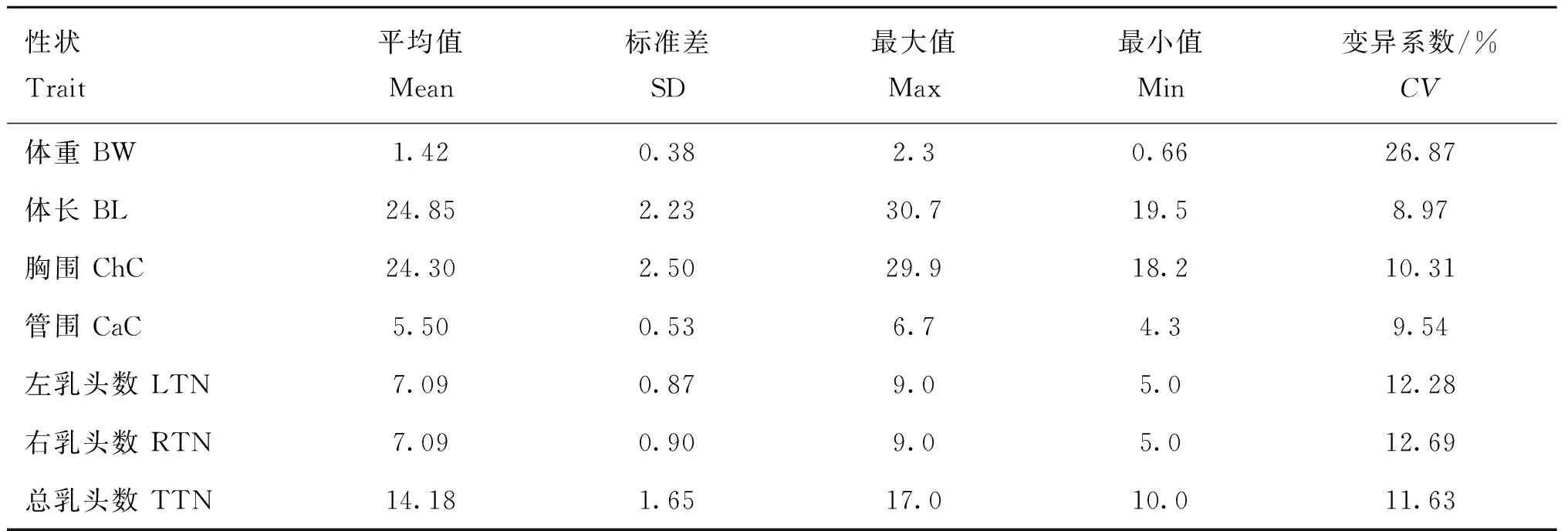
表1 苏山猪初生体尺和乳头数性状表型值的统计结果Table 1 Statistical results of phenotypic values of body conformation at birth and teat number traits in Sushan pigs
2.2 基因型填充准确度
经过利用Beagle软件对269头苏山猪的55K芯片数据进行基因型填充,共获得31 123 761个SNPs,剔除MAF<0.01的SNPs后,得到8 547 616个SNPs。利用CR和correlation估算每条染色体的基因型填充准确度(图1),结果显示,平均CR为0.71,最大值和最小值分别为0.83(SSC X)和0.67(SSC 5);平均correlation为0.58,最大值和最小值分别为0.75(SSC X)和0.52(SSC 5),基因型填充后,显著增加了55K芯片的密度(图2)。通过call rate<0.1、MAF<0.01和correlation<0.8过滤掉不符合分析要求的SNPs后,最终有2 676 280个SNPs用于后续数据分析,其平均相关性为0.93。
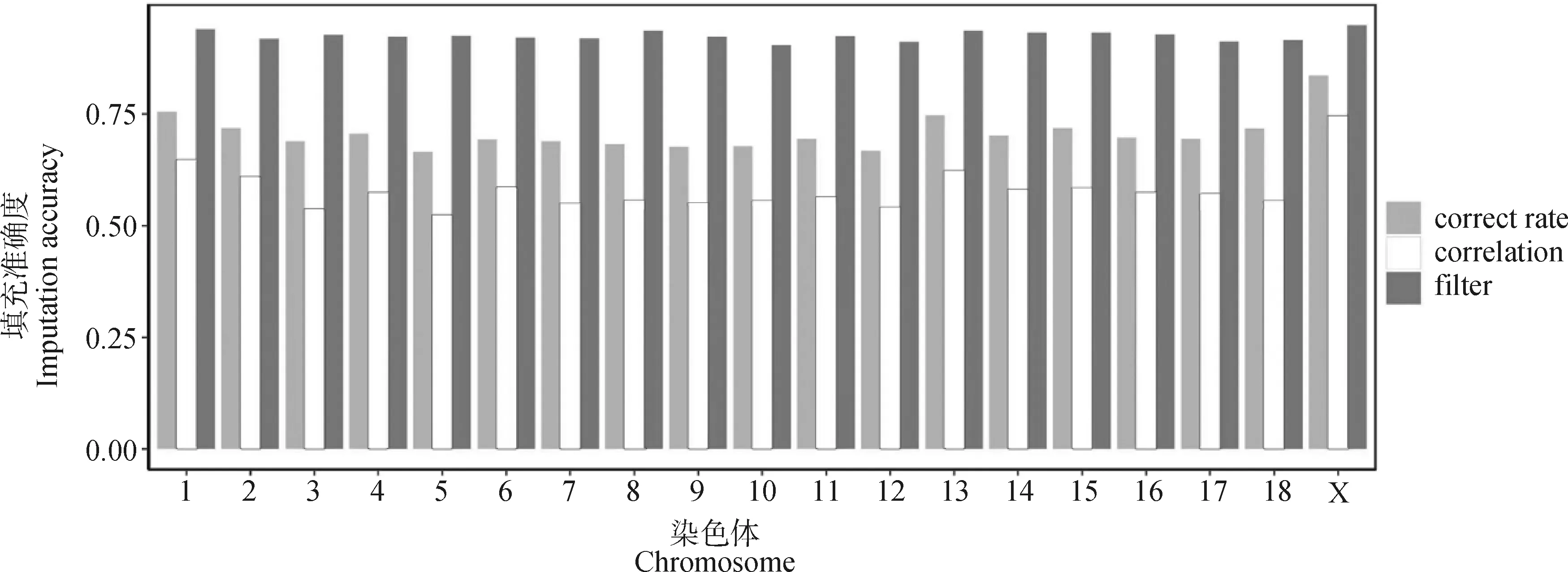
横坐标以苏山猪染色体绘制,纵坐标表示填充准确度,correct rate和correlation为过滤前的等位基因准确性和相关性,filter为过滤后的相关性The x-axis represents the chromosomes of Sushan pigs, and the y-axis represents the imputation accuracy, correct rate and correlation represent CR and correlation before filtering, filter is the correlation after filtering图1 苏山猪染色体填充准确度Fig.1 Imputation accuracy across whole chromosomes in Sushan pig population

上图和下图分别为染色体填充前和填充后的SNP密度分布,横坐标表示染色体大小,纵坐标表示染色体,刻度颜色表示1 Mb窗口内的SNP数目Above and below are figures respectively represent the SNP density distribution before and after imputation, the x-axis denotes the size of chromosomes, the y-axis denotes the chromosomes, color scale denotes the number of SNP within 1 Mb window size图2 苏山猪染色体填充前后SNP密度分布比对Fig.2 Comparison of SNP density distribution before and after imputation of chromosomes in Sushan pig population
2.3 选择信号分析和全基因组关联分析
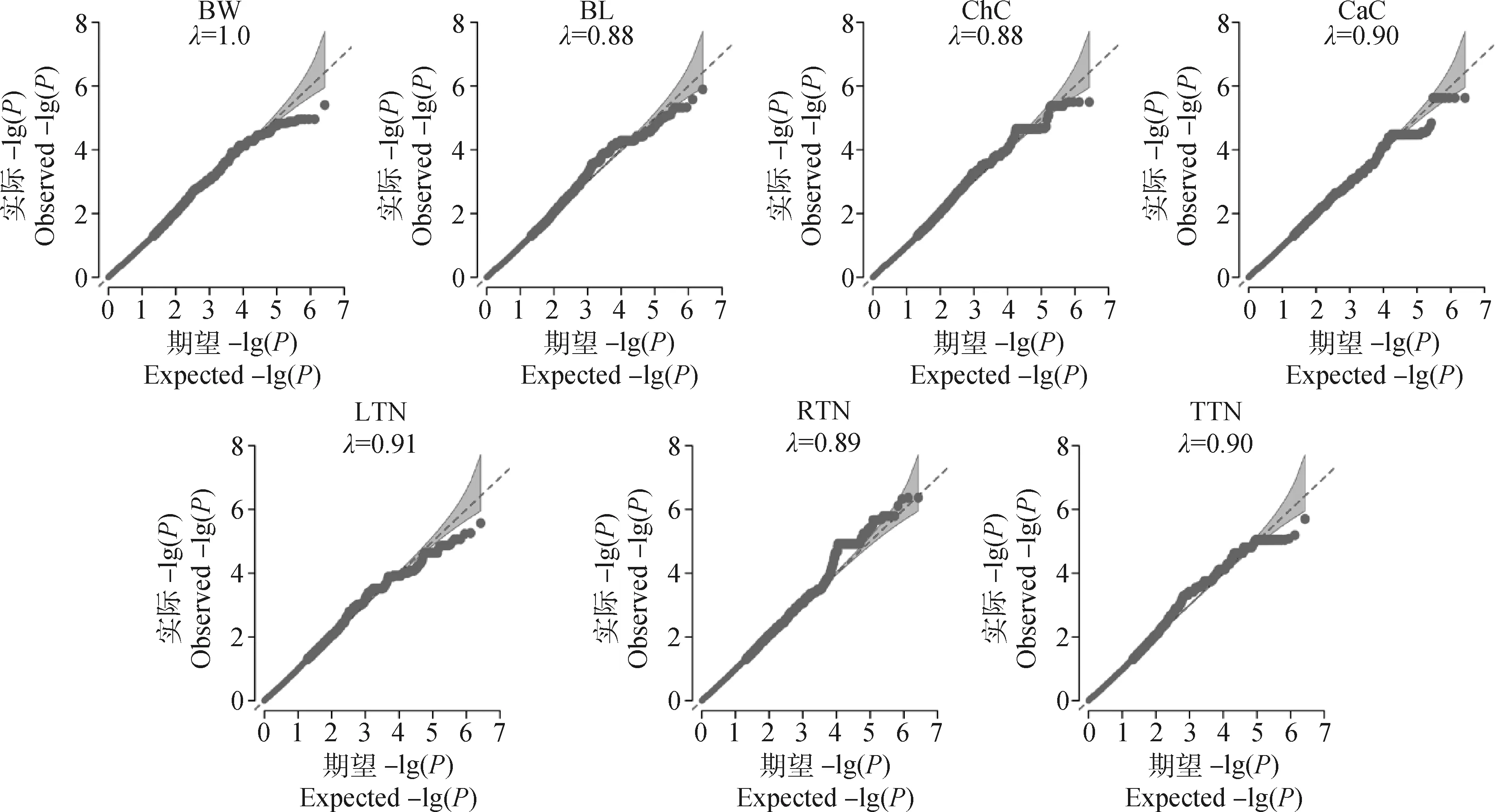
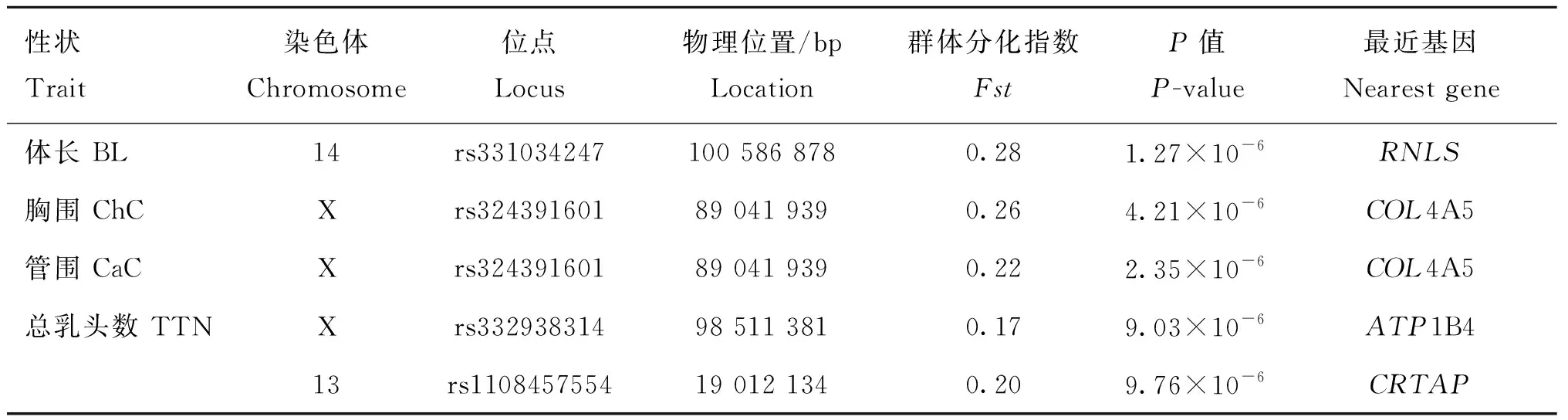
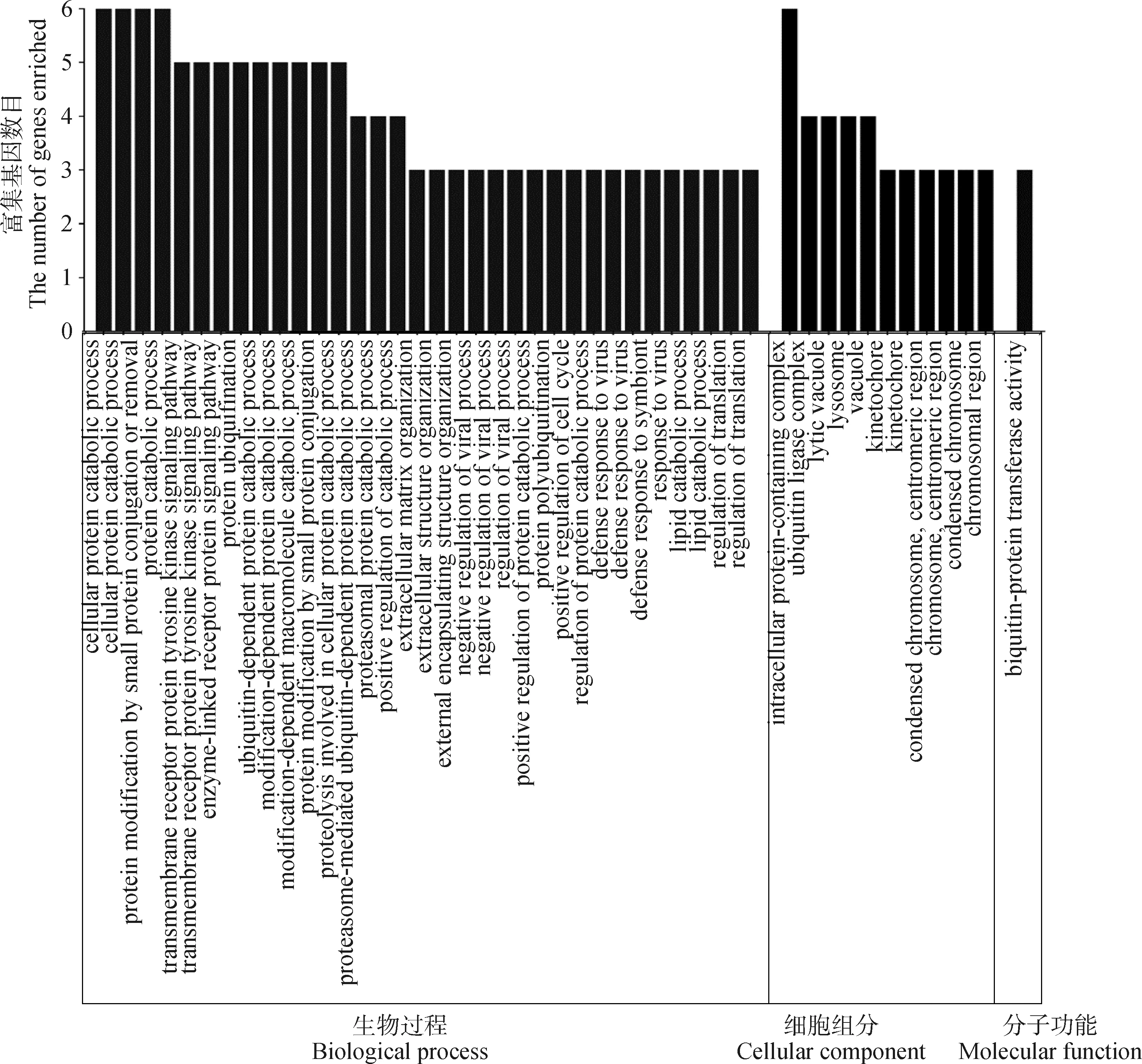
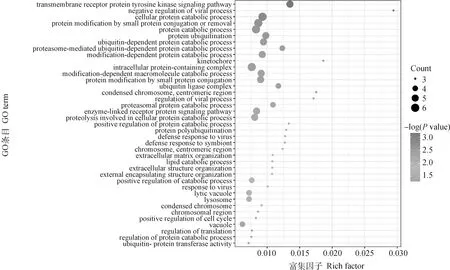
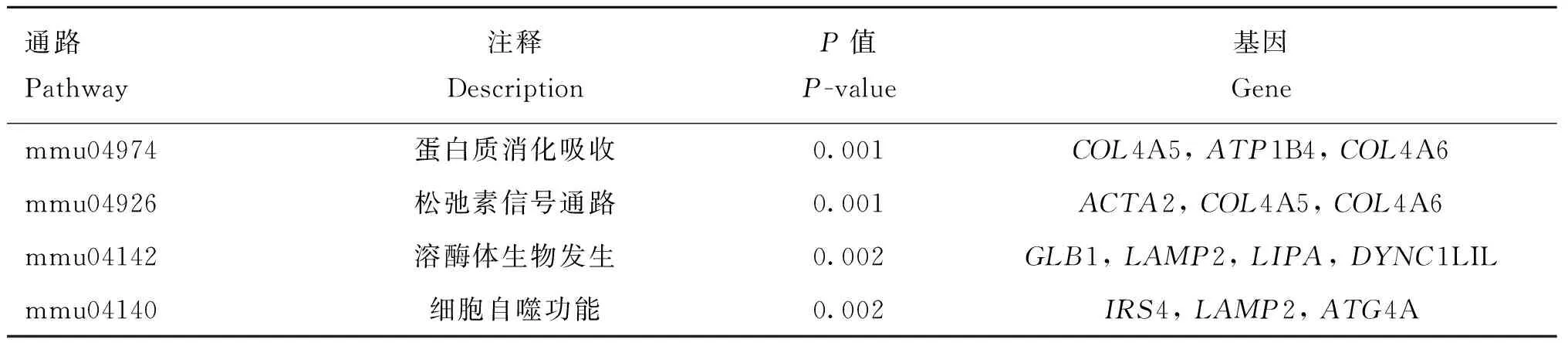
按照各性状Fst值的前1%为标准,可得体重、体长、胸围、管围、左乳头数、右乳头数和总乳头数性状的Fst阈值分别为0.16、0.20、0.20、0.17、0.14、0.14和0.15,与Wright[13]定义的高度分化的阈值范围接近(0.15 初生体尺和乳头数性状的群体分化指数和全基因组关联分析曼哈顿图进行组合。群体分化指数曼哈顿图中虚线为Top1%的阈值线,横坐标以苏山猪染色体绘制,纵坐标表示群体分化指数;全基因组关联分析曼哈顿图中横坐标以染色体绘制,纵坐标表示 -lg(P),虚线表示显著阈值线Population differentiation index and genome-wide association analysis Manhattan plots were combined for body conformation at birth and teat number traits. The dotted lines indicate the threshold line of top1% in the population differentiation Manhattan plots, the x-axis represents the chromosomes of Sushan pigs, and the y-axis represents the Fst value; genome-wide association analysis Manhattan plots are plotted with chromosomes on the abscissa, the y-axis represents the -lg(P), and the dashed lines represents significance thresholds图3 苏山猪初生体尺和乳头数性状群体分化指数和全基因组关联分析曼哈顿图Fig.3 Manhattan plots of fixation index and genome-wide association analysis for body conformation at birth and teat number traits in Sushan pig population 苏山猪群体初生体尺和乳头数性状的GWAS分析结果显示,与体尺性状显著关联的SNP有32个,乳头数性状显著关联的SNP有90个。其中,与体重显著关联的SNP有1个,位于X染色体;与体长显著关联的位点有13个,全部位于14号染色体;与胸围显著关联的位点有9个,分别位于14和X染色体;与管围显著关联的位点有9个,位于X染色体;与左乳头数显著关联的位点有6个,分别位于1、13和16号染色体;与右乳头数显著关联位点有53个,分别位于1和13号染色体;与总乳头数显著关联的位点有31个,分别位于13、16和X染色体。QQ-plot分析结果见图4,图中膨胀系数(λ)介于0.884~1.062之间,表明本研究试验群体中不存在明显的种群分层现象。 λ为基因组膨胀系数λ represents coefficient of expansion图4 苏山猪初生体尺和乳头数性状分位数-分位数图Fig.4 The quantitle-quantitle plots for body conformation at birth and teat number traits in Sushan pig population 通过对苏山猪群体初生体尺和乳头数性状进行Fst和GWAS分析,筛选出与体长、胸围、管围和总乳头数性状显著关联的4个候选位点,定位在13、14和X染色体上(表2,图3)。与体长性状显著相关的候选位点位于14号染色体,ACTA2基因为位置功能候选基因。与管围和胸围性状都显著相关的候选位点位于X染色体上,COL4A5和COL4A6基因为位置功能候选基因。总乳头数性状显著相关的候选位点分别在13和X染色体上,ATP1B4、UBE2A、NKRF、SEPTIN6、NKAP、ZBTB33、CRTAP、TRIM71、FBXL2基因为位置功能候选基因。 表2 苏山猪初生体尺和乳头数性状显著相关SNPsTable 2 The SNPs significantly associated with body conformation at birth and teat number traits in Sushan pigs 本研究基于Metascape在线网站中的小鼠数据库开展了 GO和KEGG功能富集分析。共富集到了46条显著GO条目(图5),其中参与生物过程的GO条目为34条,参与形成细胞成分的GO 条目为11条,能解释分子功能的GO条目为1条。图6显示了基因富集GO 条目的显著性。候选基因的KEGG通路富集分析中发现4条通路与蛋白质消化、松弛素信号通路、溶酶体生物发生以及细胞自噬功能有关(表3)。 横坐标表示GO条目;纵坐标表示GO条目富集的基因数The x-axis represents GO term; the y-axis represents the number of genes enriched by GO term图5 候选基因GO富集分析柱状图Fig.5 Histogram of GO enrichment analysis of the candidate genes 横坐标表示rich factor,rich factor 为位于该通路的差异表达基因个数/位于该通路的所有注释基因个数;纵坐标表示GO 条目The x-axis represents the rich factor, rich factor indicate the number of differentially expressed genes in this pathway/the number of all annotated genes in this pathway; the y-axis represents the GO terms图6 候选基因GO富集分析气泡图Fig.6 Bubble plot of GO enrichment analysis of the candidate genes 表3 苏山猪初生体尺和乳头数性状KEGG分析显著富集Table 3 Significant kyoto encyclopedia of genes and genomes (KEGG) pathway associated with body conformation at birth and teat number traits in Sushan pig population 本研究利用387头猪的重测序数据对269头猪已有的SNP芯片数据进行填充,得到更高质量和更高密度的基因数据集,以CR和相关性作为基因型填充准确性的评估指标[14]。据研究表明,影响填充准确性的因素主要包括参考群体大小[15]、群体结构[16]、最小等位基因频率[17]、填充群体之间的遗传距离[18]和填充方法[19]。Ye等[16]在对鸡的研究中使用60K数据对基因组序列进行填充,填充的准确性为0.62。Van Binsbergen等[20]在对奶牛的研究中使用基因型填充手段,填充的准确性范围分别为0.77~0.83、0.37和0.46。本研究与先前两个研究的填充准确性较为接近,表明本研究填充后的数据可用于后续分析。 先前对猪乳头数的数量基因座(QTL)研究过程中,除了3、4、5、18和19号染色体上没有发现与乳头数相关的QTL外,其余的染色体上均有QTL存在,有趣的是,在猪QTL库中发现和乳头数相关的QTL与胸椎数的QTN存在一些重叠[21-23]。Duijvesteijn等[24]发现,乳头数的QTL包含了与椎体发育相关的基因,从而解释了乳头数和脊椎数之间存在相关的生物机制[25],乳头数和脊椎数呈正相关,并且猪体长性状与胸椎数和脊椎数密切相关,体长性状相关联的基因很可能会影响猪乳头数[26-27]。rs331034247位于ACTA2基因附近,与体长性状显著关联。根据先前报道,Weigand等[28]利用正常女性和患有乳腺癌女性的乳腺细胞,分别进行培养并开展一系列表达分析,发现ACTA2与脂肪干细胞(adipose derived stem cells,ADSC)和乳腺上皮细胞(mammary epithelial cells,MES)之间存在正相关[29]。黄京书和熊远著[30]利用RT-PCR探究了ACTA2基因在多个品种猪的骨骼肌表达分析,结果显示处于不同发育阶段的骨骼肌中基因的表达量存在较大差异,ACTA2基因很可能与骨骼肌发育存在一定的相关性。由上可知,ACTA2基因很可能是体长和乳头数的候选基因。 rs1108457554位于CRTAP基因的蛋白编码区域以及FBXL2基因的下游,与TTN性状显著相关。Tang等[31]、Grol等[32]和Tonelli等[33]在人类、小鼠和斑马鱼的研究中发现,CRTAP基因中存在导致隐性成骨发育不全症的致病杂合致病突变,由此推断CRTAP基因也可能会导致猪椎体发育异常,从而影响乳头数量。FBXL2基因在调节骨骼成肌细胞增殖和成肌分化中发挥着重要作用,该基因的沉默会导致骨骼肌中的成肌细胞增殖[34-35],骨骼肌的发育异常很可能会引起动物椎骨畸形和体尺特征较差,进而导致乳头数减少和死亡率增加。在rs1108457554的下游搜寻到TRIM71 基因,Connacher和Goldstrohm[36]发现TRIM71 基因促进干细胞增殖,诱导成纤维细胞重编程为多功能干细胞,也有研究表明[37]该基因与骨骼肌的发育有关。 rs332938314与TTN性状显著相关,位于SEPTIN6、UBE2A、NKRF和ATP1B4基因的附近。Li等[38]研究发现,SEPTIN6 基因是影响氨基酸介导的奶牛乳腺上皮细胞生长和酪蛋白合成的关键积极调控因子,SEPTIN6 基因参与细胞的生理过程,包括胞质分裂、囊泡运输、细胞形态、细胞运动和细胞周期[39]。Fingar等[40]研究发现,氨基酸可以影响细胞增殖和牛奶的生成,由此推断该基因很可能与猪的乳头数以及泌乳能力存在一定关联。在对人的研究中发现,UBE2A和NKRF基因发生突变或者缺失会导致乳头间距增大、智力障碍、全身多毛症和泌尿生殖系统发育异常等症状[41-43],这两个基因突变和缺失是否会导致猪出现相同的症状,还需要开展相关研究。Pestov等[44]在对小鼠的研究中发现,ATP1B4基因表达量会对表皮的生长产生影响,在其他研究中发现ATP1B4基因编码的蛋白质βm主要在骨骼肌中高水平表达[45-47]。 rs324391601位于COL4A5基因内和COL4A6基因上游附近,与ChC显著关联。Jiang 等[48]在对湖羊的研究中发现,COL4A6基因是ChC的候选基因,本研究中对苏山猪胸围性状候选基因搜寻中也得出相同的结果,因此COL4A6基因可以作为ChC的候选基因。在对日本和牛的研究中发现,COL4A5基因在肌内脂肪组织中高特异性表达,与日本和牛的肌内脂肪形成有关,可以将该基因作为猪肌内脂肪研究的相关基因。 富集分析结果表明,ACTA2、CRTAP、TRIM71、COL4A5和COL4A6基因主要显著富集在跨膜受体蛋白酪氨酸激酶信号通路,该通路可以影响乳腺癌细胞增殖和凋亡[49]。ACTA2、COL4A5和COL4A6基因同时显著富集在松弛素信号通路,通路异常可以影响小鼠的乳头严重发育不良,造成无法哺乳后代[50]。结果表明,这些候选基因可能在猪胚胎发育过程中对体形态和乳头数性状起重要作用。 本研究基于SNP数据集进行了Fst和GWAS分析,对显著性SNPs进行注释和富集分析之后,发现了1个与体长性状相关的候选基因ACTA2;发现了1个与胸围性状相关的候选基因COL4A6;发现了3个与乳头数性状相关的候选基因ACTA2、CRTAP和SEPTIN6,为苏山猪体尺性状和乳头数性状的遗传改良提供了新的视角。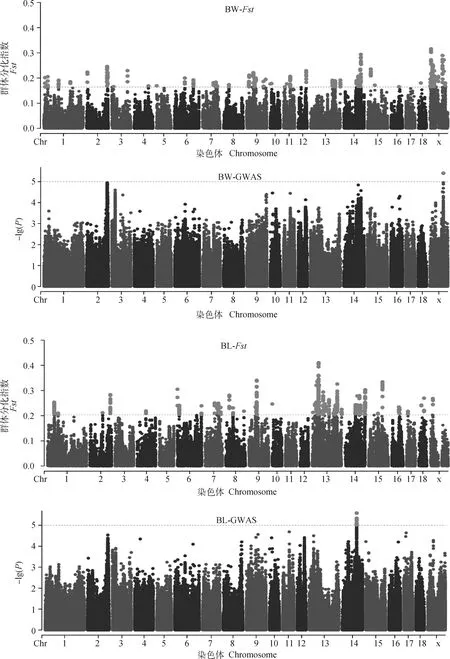
2.4 富集分析
3 讨 论
4 结 论
