特殊类型肾上腺疾病2例报告
2023-02-02袁雪雯楚闪闪朱子阳
袁雪雯,楚闪闪,朱子阳,顾 威
南京医科大学附属儿童医院内分泌科,江苏 南京 210008
原发性肾上腺皮质功能减退症(primary adrenal insufficiency,PAI)是指肾上腺皮质本身病变所导致的皮质激素合成减少。临床病因较多,主要包括遗传性因素,如先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)、先天性肾上腺发育不全等,及其他免疫、感染、代谢、肿瘤等导致的肾上腺皮质功能不全。临床一般表现为糖皮质激素分泌不足或作用缺陷,伴或不伴盐皮质激素及性激素分泌异常,很少只表现一种激素缺乏。家族性糖皮质激素缺乏(familial glucocorticoid deficiency,FGD),亦称为促肾上腺皮质激素(adrenocorticotropic hormore,ACTH)不敏感综合征,肾上腺皮质对ACTH不敏感,导致孤立性糖皮质激素合成不足,而无盐皮质激素及性激素分泌异常。醛固酮合成酶(aldosterone synthase,CYP11B2)基因突变导致醛固酮生物合成代谢途径的最后步骤被破坏[1],引起醛固酮合成酶缺乏症,临床表现为孤立性盐皮质激素缺乏,而无糖皮质激素及性激素合成异常。本院2例是临床罕见的肾上腺疾病,均为常染色体隐性遗传性疾病,迄今国内对相关病例的报道比较少。本文将对2例特殊类型肾上腺疾病的临床特点、诊治过程、基因结果进行回顾性分析,同时进行临床资料总结,以提高临床医师对该病的认识。
1 临床资料
患儿1,女,5 岁3 个月,因“发现皮肤变黑5 年余”就诊。患儿出生后即出现皮肤变黑,但无反复各系统感染,无频繁恶心、呕吐、腹泻等表现,2年多前就诊于当地医院,查ACTH 增高,皮质醇减低,予以激素替代治疗,皮肤颜色有好转,但近期再次出现皮肤变黑来本院就诊。患儿为母亲第1 胎第1产,足月顺产,出生体质量2.95 kg,否认围生期缺氧窒息史等。父母均体健,非近亲结婚,有1弟弟3岁余,体健,家族成员中无类似疾病史。体格检查:身高123.7 cm(位于同年龄、同性别、同种族儿童生长曲线的第97 百分位数以上),体重25 kg,血压94/67 mmHg,全身皮肤黝黑,唇部、牙龈、关节等褶皱部位显著(),双侧乳房B1期,心肺腹未见异常,外阴女童式,未见阴毛腋毛生长。辅助检查(括号内为正常值):血、尿、粪便常规及肝肾功能、电解质均无异常。皮质醇18.56 nmol/L(133~537 nmol/L),ACTH>2 000 pg/mL(0~46 pg/mL);孕酮<0.159 nmol/L(0~0.474 nmol/L);睾酮<0.087 nmol/L(<0.087 nmol/L);17α-羟孕酮<0.05 ng/mL(<2.32 ng/mL);硫酸脱氢表雄酮0.25 μg/mL(2.8~85.2 μg/mL);雄烯二酮<0.3 μg/mL(≤4.01 ng/mL);血浆肾素活性:2.36 ng/(mL·h)[1.45~5.00 ng/(mL·h)],血管紧张素Ⅰ:1.629 μg/L(无参考范围),血管紧张素Ⅱ:73.08 ng/L(32~90 ng/L),醛固酮:83.61 ng/L(50~313 ng/L);染色体46,XX。左腕关节正位片(骨龄片)相当于5 岁。肾上腺CT 平扫提示左侧肾上腺未见明显异常,右侧肾上腺显示欠佳。垂体MRI 平扫显示蝶鞍形态正常,所见蝶骨信号无异常。垂体高度为5.1 mm,前后叶信号未见异常,垂体柄居中无偏斜。视交叉无移位,信号无异常。鞍上池、桥前池及三脑室漏斗部未见填塞及扩张。头颅MRI 平扫显示未见明显异常。
患儿2,男,1个月16 d,因“呕吐1月余”入院,出生后1 周无明显诱因下出现反复呕吐,呕吐物为胃内容物,伴体重减轻,多次查电解质提示低钠、高钾收住我科。患儿为母亲第3胎第3产,足月剖宫产,出生为超重儿(体重4.25 kg),身长不详,无宫内窘迫史,出生后无窒息。父母体健,否认近亲婚配,第1 胎出生后因“先天性食道闭锁”夭折,有1 个姐姐6 岁余,体健,否认家族性遗传疾病史。体格检查:体重3.9 kg,神清,精神反应一般,皮下脂肪菲薄,皮肤黏膜色泽正常,前囟、眼窝凹陷,心肺腹未见异常,双侧睾丸容积2 mL,阴茎长3 cm(处于同龄儿正常范围内),阴囊无色素沉着,四肢活动正常。辅助检查(括号内为正常值):电解质:血钠110.8 mmol/L(135~145mmol/L),血氯75.6mmol/L(96~108mmol/L),血钾6.65 mmol/L(3.5~5.5 mmol/L);肝功能、肾功能未见异常;血尿代谢筛查未见异常;甲状腺功能7项未见异常;皮质醇252.5 nmol/L,ACTH 18.26 pg/mL;性激素:E2 <18.35 pmol/L(41.4~159 pmol/L),孕酮1.86 nmol/L,睾酮11.94 nmol/L,PRL 1 270 mU/L(86~324 mU/L),FSH 3.19 mU/mL(1.5~12.4 mU/L),LH 7.42 mU/L(1.7~8.6 mU/L);基础17α-羟孕酮、硫酸脱氢表雄酮、雄烯二酮均正常范围;血浆肾素活性:1.3 ng/(mL·h),血管紧张素Ⅰ:0.828 μg/L,血管紧张素Ⅱ:77.51 ng/L,醛固酮:95.86 ng/L;染色体46,XY。胸腹立位片:两肺纹理增多。上消化道造影:胃扭转。心电图:窦性心动过速。心脏彩超B 超:卵圆孔未闭,心功能未见异常。腹部(肝胆胰脾肾上腺)B 超:未见异常。双侧腹股沟睾丸B 超:未见异常。肾上腺CT平扫:两侧肾上腺稍增粗。垂体MRI平扫未见异常。头颅MRI平扫:两侧额顶颞部脑外间隙增宽(左侧明显);两侧脑室前角旁软化灶可能;左侧侧脑室较右侧略宽(图2)。
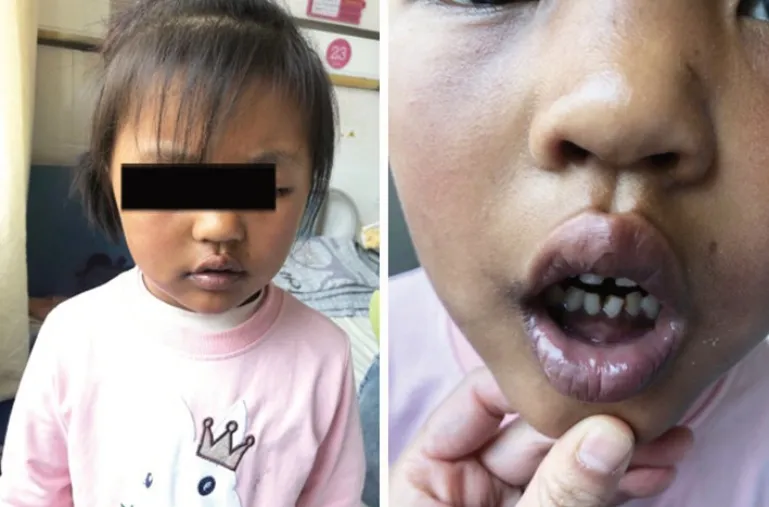
图1 病例1患儿
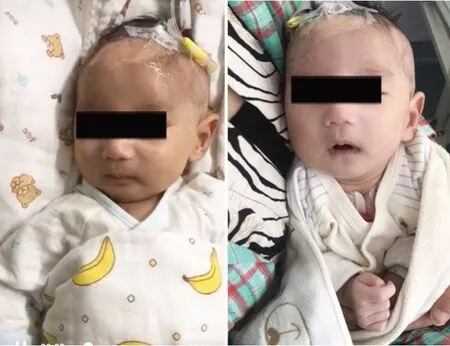
图2 病例2患儿
在治疗过程中,经家属签署知情同意书后,分别留取患儿1、患儿1父母和弟弟及患儿2、患儿2父母和姐姐的外周血标本各2 mL,EDTA 抗凝。DNA提取、PCR 产物电泳、纯化、测序及序列分析由北京迈基诺医学检验所进行。基因测序结果显示:患儿1 MC2R 基因外显子区域存在复合杂合突变:c.433C >T(p.R145C)和c.712C >T(p.H238Y)。MC2R 基因的第145 号氨基酸由精氨酸变为半胱氨酸和第238 号氨基酸由组氨酸变为酪氨酸,经过Sanger测序确认(图3),该变异位点分别来自父亲和母亲,均为已报道的致病变异,患儿弟弟携带与母亲相同的变异基因型。患儿2 CYP11B2基因外显子区域发现存在复合杂合突变:c.240-1G>T(splicing)和c.1342C >T(p.R448C)。CYP11B2 基因的第1 342 号核苷酸由胞嘧啶变为胸腺嘧啶和第448 号氨基酸由精氨酸变为半胱氨酸,经过Sanger 测序确认(图4),该变异位点分别来自父亲和母亲,均未有相关报道,患儿姐姐携带与母亲相同的变异基因型。
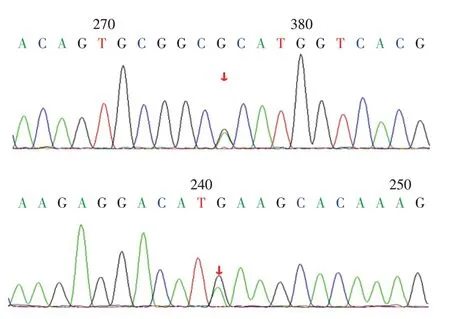
图3 病例1家族性糖皮质激素缺乏患儿MC2R基因外显子区域存在复合杂合突变
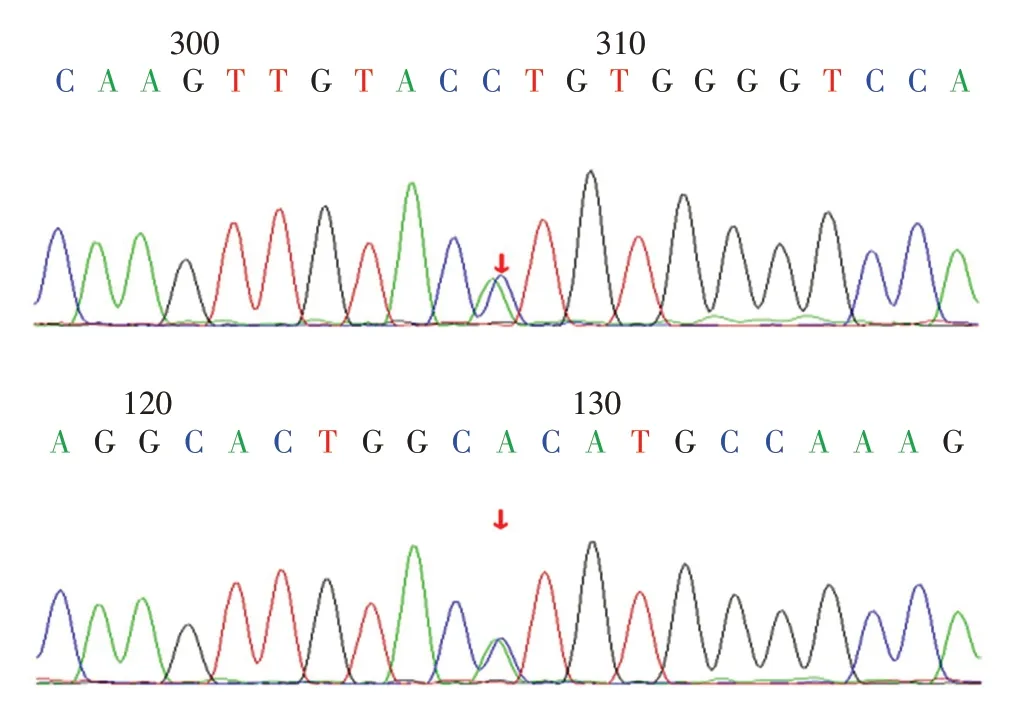
图4 病例2 醛固酮合成酶缺乏症患儿CYP11B2 基因外显子区域存在复合杂合突变
治疗及随访:患儿1 入院后存在肾上腺皮质功能减退,给予静脉琥珀酸氢化可的松替代治疗,好转后逐渐改成口服激素治疗,在减量过程中出现ACTH 及皮质醇波动,但电解质水平正常,予加用9α-氟氢可的松0.05 mg 每天2 次口服,复查各项指标正常,皮肤色素沉着明显好转,予以带药出院,出院时口服醋酸氢化可的松10 mg 每8 h 1 次[约31 mg/(m2·d)],9α-氟氢可的松0.05 mg每天2次。出院后随访,基因结果提示只存在糖皮质激素合成障碍,故逐渐停用盐皮质激素,目前醋酸氢化可的松用量为2.5 mg 每8 h 1 次[约7.5 mg/(m2·d)],患儿肾上腺激素水平控制尚可。患儿2有明显高钾低钠电解质紊乱,考虑肾上腺危象,予静脉补液及琥珀酸氢化可的松治疗,期间复查电解质有好转,但激素减量过程中再次出现高钾低钠,且患儿ACTH 及皮质醇水平一直在正常范围,无性激素水平异常,虽然检测醛固酮水平不低但患儿失盐的临床表现明显,且为补液后所检测,故不排除患儿醛固酮出现了问题,加用9α-氟氢可的松0.05 mg每天2 次口服,但电解质紊乱纠正不明显,加量至9α-氟氢可的松0.1 mg每天2次后电解质逐渐恢复到正常范围,基因结果证实醛固酮合成障碍,遂逐渐减量糖皮质激素至停用,现出院后患儿无呕吐症状,食纳佳,体重增长明显,复查患儿电解质水平稳定。
2 讨论
儿童肾上腺疾病病因复杂,临床表现多样化,临床诊断及鉴别诊断困难,常需要借助分子诊断明确病因。本文描述的两类肾上腺疾病比较特殊,只表现为单一肾上腺激素缺乏,与既往常见肾上腺疾病不同,临床罕见。
患儿1 临床以皮肤黑为主要表现,无呕吐等失盐症状,也无性腺发育异常,完善相关检查发现患儿肾上腺激素合成通路中只存在糖皮质激素合成异常,完善基因信息后确诊为FGD。该疾病是一种罕见的常染色体隐性遗传病,由Shepard 等[2]在1959 年首次报道,国外报道较多,国内鲜有报道。肾上腺皮质对ACTH 不敏感,导致孤立性糖皮质激素合成不足,血清皮质醇低而ACTH明显增高,临床表现为低血糖、反复感染、皮肤色素沉着、身材高大等特点,无失盐表现。本文中患儿自出生后起病,有明显的皮肤色素沉着,但是无低血糖发作及反复感染病史,与既往报道不一致。患儿出现身材高大,一方面可能与过高的ACTH 刺激黑色素受体在骨和软骨中表达,从而刺激骨和软骨生长,另一方面与糖皮质激素缺乏,降低了胰岛素样生长因子结合蛋白5(insulin-like growth factor-binding protein-5,IGFBP-5)对骨生长的抑制作用有关[3-4]。在1993 年首次报道FGD 相关基因突变[5],目前已报道的相关基因有黑皮质素2受体(MC2R)、黑皮质素2受体辅助蛋白(MRAP)、烟酰胺核苷酸转氢酶(NNT)、微小染色体修复缺陷4同源基因(MCM4)、硫氧还蛋白还原酶2(TXNRD2)、类固醇激素急性调节蛋白(STAR)、细胞色素P450(家族11,亚家族A,多肽1)(CYP11A1)及特殊类型三A 综合征(AAAS)相关基因[6]。本文报道的是MC2R突变所致FGD的发生,该基因又称ACTH 受体基因,定位于18p11.2(OMIM 607397),是一种G蛋白耦联的跨膜受体[7],与ACTH结合直接发挥生物学效应。目前已报道40 余种致病突变,其中以点突变为主,无义突变和移码突变少见,大多数突变导致MC2R不能转运至细胞膜,少数突变导致MC2R 不能与ACTH 结合[8]。例1 患儿经基因检测亦发现为错义变异,且致病性变异已有报道[9-10],根据2015 年美国医学遗传学与基因组学(ACMG)指南,该变异为可能致病性变异。
患儿2 以呕吐为主要临床表现,且存在高钾低钠电解质紊乱,但皮质醇正常,醛固酮水平不高。在治疗过程中,糖皮质激素减量后再次出现高钾低钠,予口服补充盐皮质激素后电解质水平恢复正常,考虑醛固酮可能存在合成不足或醛固酮抵抗,但多次复查醛固酮水平不高,故锁定醛固酮合成出现了问题,完善基因后发现醛固酮合成酶(aldosterone synthase,CYP11B2)存在复合杂合突变c.240-1G >T(剪接突变)和c.1342C >T(错义突变),且国内外无该位点的报道,最终确诊为醛固酮合成酶缺乏症,提示该患儿只存在盐皮质激素合成障碍。该疾病是CAH 的一种极为罕见的特殊类型,为常染色体隐性遗传性疾病,是由位于8号染色体长臂上的醛固酮合成酶CYP11B2 基因突变所致[11-12],从而导致醛固酮合成酶活性缺乏或降低,影响了醛固酮的合成代谢途径。临床表现有反复呕吐、腹泻、脱水、体重不增等盐皮质激素缺乏的症状,实验室检查有低钠低氯高钾血症,代谢性酸中毒,高肾素低醛固酮血症。发病年龄越小症状越明显,随着年龄的增长,一方面从食物中摄入的钠增加,另一方面肾小管对盐皮质激素耐受性下降,临床表现可逐渐好转[13-14]。此类疾病因临床罕见需与假性醛固酮减少及其他类型肾上腺疾病相鉴别,避免造成误诊漏诊。
对于肾上腺疾病的治疗,均采用激素替代治疗。病例1 患儿只需进行糖皮质激素终身替代治疗,剂量遵循个体化原则,一般无需补充盐皮质激素。儿童首选醋酸氢化可的松,以保证儿童正常生长发育,剂量为10~12 mg/(m2·d)。病例2主要是口服或静脉补钠及盐皮质激素(9α氟氢可的松)替代治疗,剂量为0.05~0.10 mg/d,严重失盐者可增至0.20 mg/d。治疗过程中需监测电解质及血压情况,及时调整用药剂量。
综上所述,肾上腺疾病病因复杂,表现形式多样,孤立性一种激素缺乏临床罕见,单纯依据临床表现明确其病因有一定难度,确诊还需依靠基因检测。临床医生需增加对该类疾病的认识,以期早期诊断、早期治疗。
