1,4-丁二醇在m-ZrO2(11)面吸附活化机理第一性原理研究
2023-01-31吴文海缪长喜姜冬宇樊志贵
吴文海,缪长喜,姜冬宇,樊志贵,宋 磊
(中国石化 上海石油化工研究院,上海 201208)
3-丁烯-1-醇(3-buten-1-ol,BTO)是一种高附加值的不饱和醇,由于分子结构中同时具有双键和羟基,性质活泼,可参与多种类型的反应,用于医药、农药中间体、聚合物功能材料以及食物添加剂等领域,尤其是用于抗肿瘤药物、抗艾滋病药物的重要中间体的合成[1]。相比采用传统的丙烯甲醛加成法、3,4-环氧-1-丁烯还原法、3-丁烯酸还原法、3-丁炔-1-醇加氢法得到3-丁烯-1-醇,采用1,4-丁二醇(BDO)作为原料,通过气相选择性脱水来获得3-丁烯-1-醇产品的工艺路线,反应式如下:
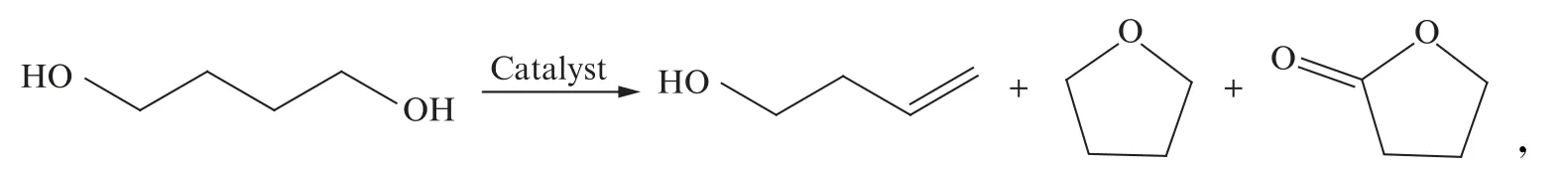
可一步得到丁烯醇产品,反应过程更加高效,并且最大限度减少了副产物,是一个绿色、原子经济性高的反应过程,受到了广泛的关注[2-3]。
如上述反应式所示,丁二醇分子在催化剂作用下,选择性脱水生成丁烯醇,主要副产物是四氢呋喃和γ-丁内酯。已经发现多种类型的金属氧化物可以用作丁二醇选择性脱水的催化剂,既可以是氧化还原特性的金属氧化物,如稀土氧化物等[4-6],也可以是具有酸碱特性的氧化物[7]。氧化锆(ZrO2)是一类具有酸碱特性的催化剂,研究表明氧化锆表面的选择性脱水与催化剂表面酸、碱中心密度相关[7-8],催化剂表面的酸碱特性对1,4-丁二醇选择性脱水具有重要的意义,在气相脱水反应过程中引入碱性气体抑制酸性位点,可以提高目标产物的选择性;而引入酸性气体则会抑制碱性位点活性,导致目标产物选择性明显降低[9]。
不同晶相的氧化锆表面可以催化低碳醇分子发生不同的反应。低碳醇分子的羟基可以采用不同形式吸附在四方相的氧化锆表面,在不同的反应条件下可以发生脱氢、脱水、耦合等一系列复杂的表面反应[10-11],生成乙烯、丁二烯等不饱和有机物。而在单斜氧化锆表面会发生完全不同的反应,原位漫反射红外(in-situDRIFTS)研究给出了丁二醇分子羟基与Yb2O3/m-ZrO2表面作用的证据[3],丁二醇分子吸附在催化剂表面会形成不同类型的单齿吸附的丁基氧化物,并经过烯醇过渡态生成丁烯醇产物,值得注意的是,丁二醇分子在该表面并不会发生连续脱水生成丁二烯产物。密度泛函理论(Density Functional Theory,DFT)研究说明在稀土氧化物表面的丁二醇分子可能会通过两个端羟基以及β-H与氧化物表面形成三齿吸附作用机理实现脱水反应[12]。然而,目前对于丁二醇分子在锆氧化物表面吸附形式推测较多,关于1,4-丁二醇在单斜二氧化锆(m-ZrO2)表面的吸附形式、活化机制依然缺少系统的研究,这是深入理解丁二醇选择性脱水反应机理,开发高效选择性脱水催化剂的起点。
本工作采用第一性原理计算方法以及周期性超胞模型,研究1,4-丁二醇分子在单斜二氧化锆(m-ZrO2)的表面的吸附过程,通过福井函数指标来判断丁二醇分子不同位置的亲电/亲核特性以及在单斜氧化锆(11)表面的吸附位点,得到丁二醇反应分子在氧化锆表面可能的稳定吸附几何构型和吸附能,尝试说明丁二醇分子在表面的吸附位点、电子转移情况以及丁二醇分子的活化、选择性脱水的反应机理。
1 计算方法和反应机制
1.1 设计理论模型和计算方法
通过密度泛函理论研究1,4-丁二醇在单斜二氧化锆(m-ZrO2)的表面吸附活化机理。本工作中的所有计算均是由Material studios软件包的Dmol3模块完成的,体系的交换相关能采用GGA-PW91泛函来描述。采用全电子内核处理方式,所有电子都包含在计算体系中进行处理。价电子波函数采用双数值型基组加极化函数(DNP)展开,为了提高计算效率,轨道热占据采用0.005 Hartree,实空间截断半径采用5.0Å。自洽收敛过程参数能力收敛10-6a.u./Å,最大受力0.002 Ha/Å,最大位移0.005Å。
在优化二氧化锆晶胞的时候,布里渊区采用5×5×5的K 点网格来模拟。单斜二氧化锆(表示为m-ZrO2)表面采用(2×2)超晶胞,c轴方向采用3层O—Zr—O 原子重复单元的模型来模拟二氧化锆表面(见图1)。为了消除平板层与层之间的相互作用,设置的真空层高度为16,布里渊区采用2×2×1的K点网格来进行模拟。在进行结构优化时,最下面两层原子(2 层O—Zr—O 结构)固定在体相结构中,最上面一层原子以及吸附的物种在优化过程中可以被弛豫。
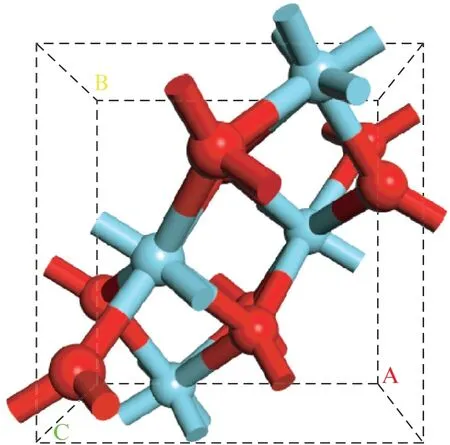
图1 单斜二氧化锆晶胞Fig.1 Crystal cell of monoclinic ZrO2
吸附能的计算公式如下:

式中:Egas-slab、Egas和Eslab分别代表分子吸附在纯净的m-ZrO2(11)表面上的总能、气相分子的能量和纯净的m-ZrO2(11)板层的能量。从式(1)可知,吸附能数值越负,代表基体与吸附分子之间具有更加强烈的相互作用。
表面模型: 二氧化锆是锆的主要氧化物,单斜二氧化锆的空间群为P21/c。采用上述计算参数优化得到的单斜二氧化锆晶格参数与实验值[13]对比见表1,表中给出的计算值与实验值能够较好地吻合。
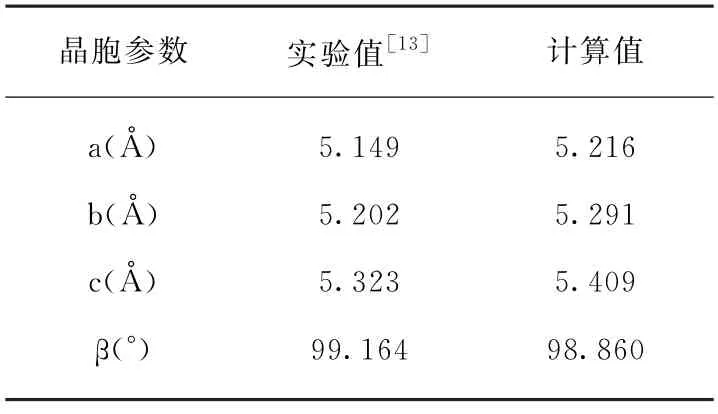
表1 单斜二氧化锆晶格参数实验值与计算值对比Tab.1 Comparison between experimental and calculated lattice parameters for monoclinic ZrO2
研究表明,单斜二氧化锆(m-ZrO2)最稳定的晶面为面,沿体相的面切出2×2×1超胞用于模拟周期性表面结构[10],如图2(770 页)所示。二氧化锆本身难以被还原,因此在本工作中不考虑氧空位的情况[14-15]。
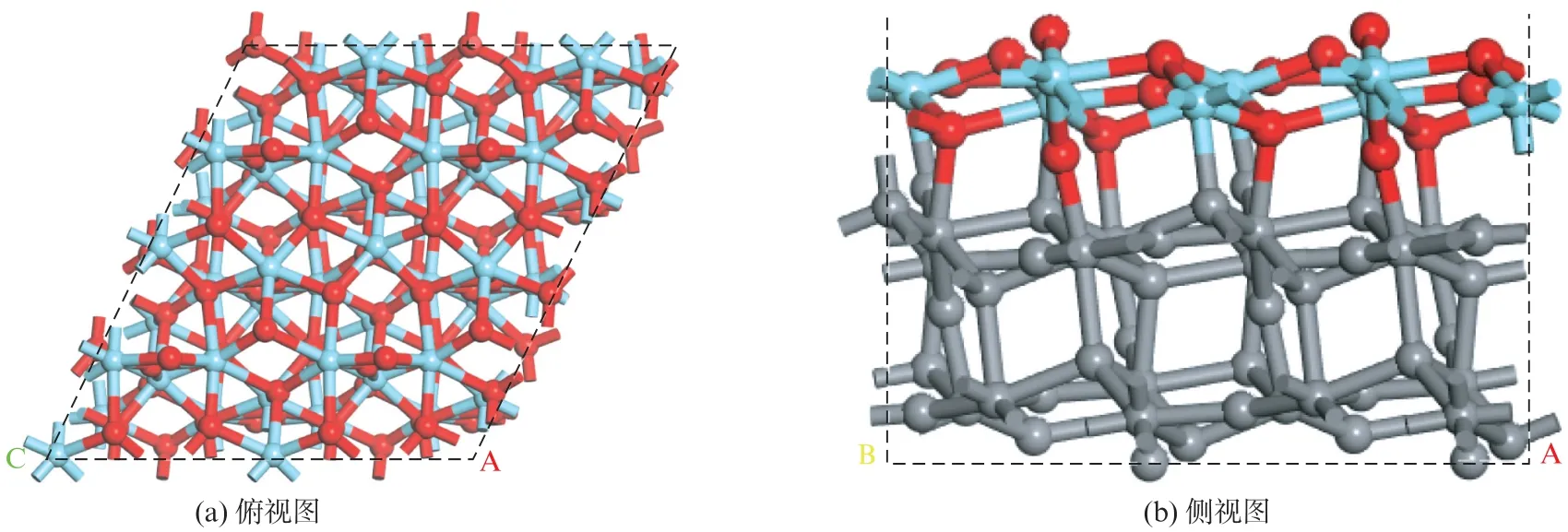
图2 m-ZrO2(11)的2×2×3表面结构Fig.2 Top and side view of the 2×2×3 supercell of m-ZrO2(11)
不同构型吸附分子在氧化锆表面β-H 脱除基元反应的过渡态通过线性同步变换(Linear Synchronous Transit,LST)/二次同步变换(Quadratic Synchronous Transit,QST)的方法搜索。
2 结果与讨论
2.1 m-ZrO2(11)面结构与电子特性
实验以及理论研究表明[16-17],m-ZrO2的(11)面是最稳定的暴露晶面。m-ZrO2(11)表面最外层原子的几何结构如图3所示。氧化锆表面存在4种锆原子(Zr(1),Zr(2),Zr(3),Zr(4)),都是六配位的锆原子。同时,表面还存在7种不同的氧原子(O(5),O(6),O(7),O(8),O(9),O(10),O(11)),其中O(5)是二配位位点,在外表面暴露更加充分,而O(6),O(7),O(8),O(9),O(10),O(11)是三配位位点。
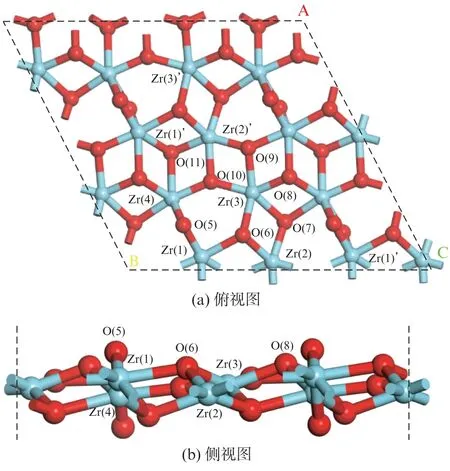
图3 m-ZrO2(11)表面结构Fig.3 Surface structure of m-ZrO2(11)
采用福井函数指标(Fukui Function Index)来分析、预测1,4-丁二醇吸附分子和单斜氧化锆表面的电子贡献-接受作用位点。每个位点分别采用亲核指数fk(-)和亲电指数fk(+)来衡量对电子的贡献和接受能力[18-19]。更高的fk(-)或fk(+)值分别意味着更强的贡献电子或接受电子的能力。表2给出了1,4-丁二醇吸附分子中不同位置的碳、氢原子和单斜氧化锆表面不同位置原子的福井指数。图4给出了1,4-丁二醇分子的结构与电子密度。
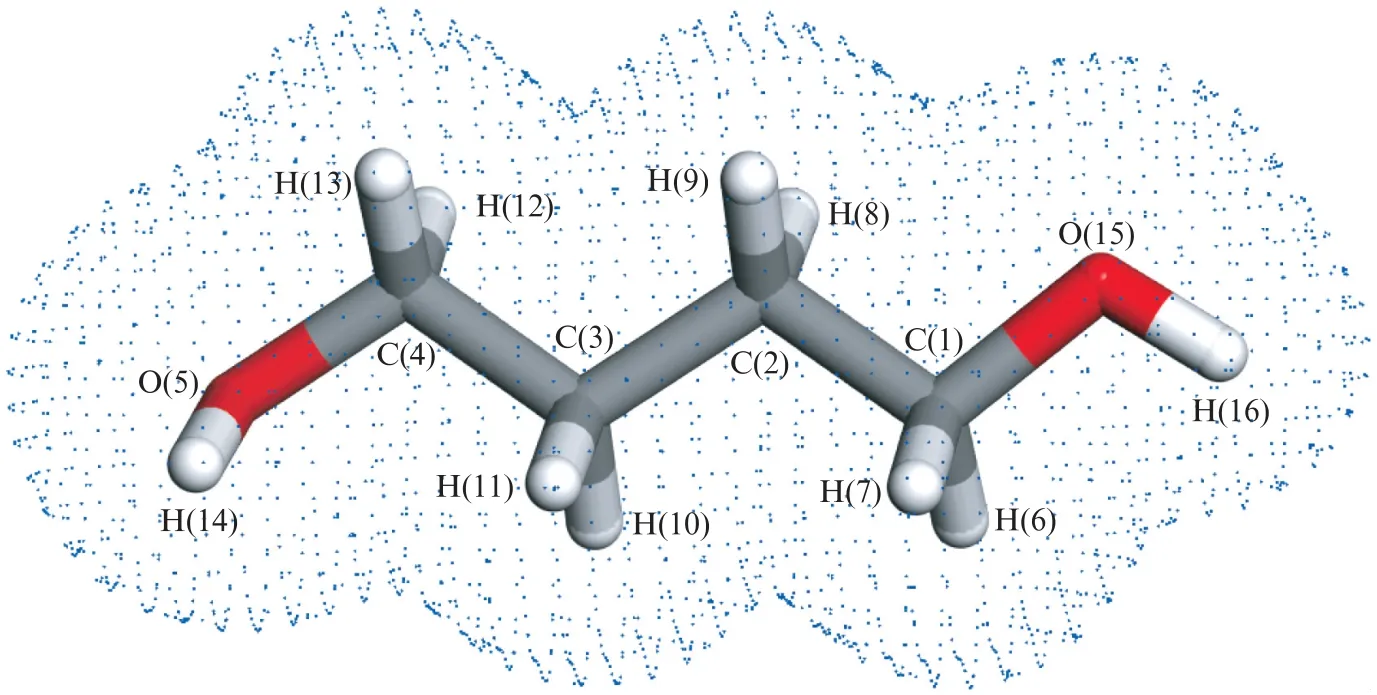
图4 1,4-丁二醇分子结构与电子密度Fig.4 Structure and electron density for 1,4-butanediol
表2 1,4-丁二醇与m-ZrO2(11)表面原子福井函数指标Tab.2 Calculated Fukui function index for 1,4-butanediol and m -ZrO2(11)surface atoms
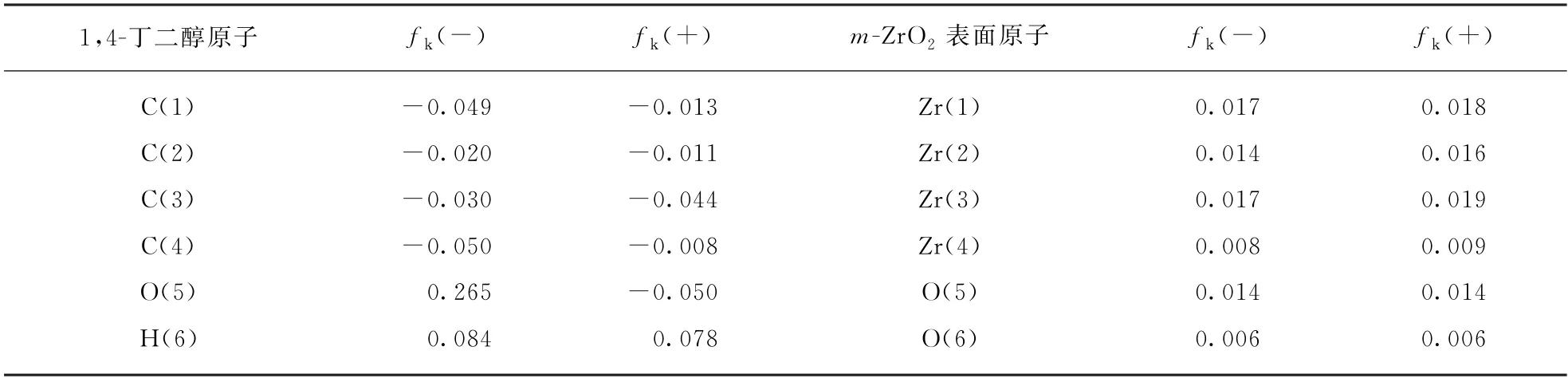
表2 1,4-丁二醇与m-ZrO2(11)表面原子福井函数指标Tab.2 Calculated Fukui function index for 1,4-butanediol and m -ZrO2(11)surface atoms
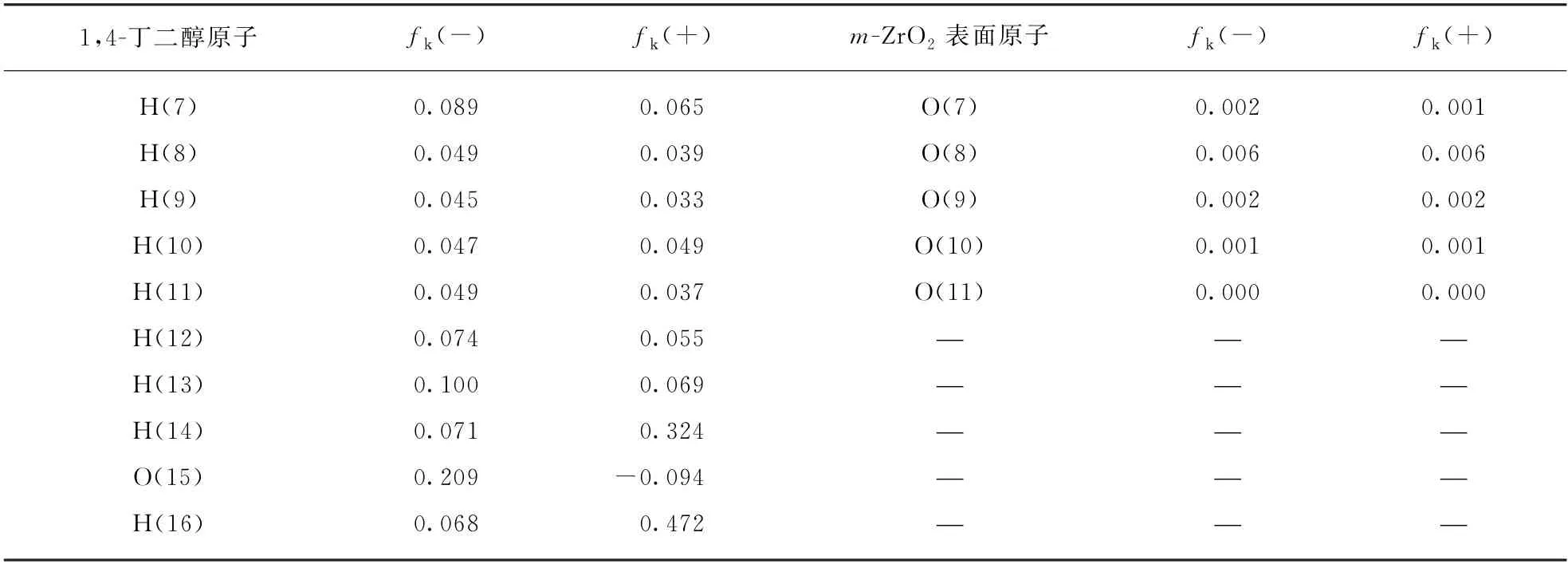
(续表)
m-ZrO2(11)表面的4种锆原子具有不同的接受电子的特性。Zr(1),Zr(2),Zr(3)适合成为接受电子中心,而Zr(4)无论是接受电子还是贡献电子的能力都弱于前者。
二配位的氧原子(O(5))的福井指标明显高于其它的三配位的氧原子(O(6),O(7),O(8),O(9),O(10),O(11)),说明O(5)的反应性能要高于其他的氧原子;O(11)的fk(-)、fk(+)值都是零,说明O(11)既不容易给出电子也不容易得到电子。此外,O(11)陷入在氧化物表面下方,从空间结构上也不容易与反应物分子上的原子接触发生反应,是相对惰性的表面氧原子。
1,4-丁二醇分子中的氧原子O(5)、O(15)具有相对高的fk(-)值,是明显的贡献电子中心,是1,4-丁二醇分子在氧化锆表面的锚定位点;而两个羟基上的氢原子H(14)、H(16)具有强的接受电子的趋势。
2.2 吸附构型优化
根据反应物以及二氧化锆表面锆原子的反应活性,分别搭建了反应物与产物在锆氧化物表面不同位点上的顶式(Top)、桥式(Bridge)吸附模型,如图5所示。
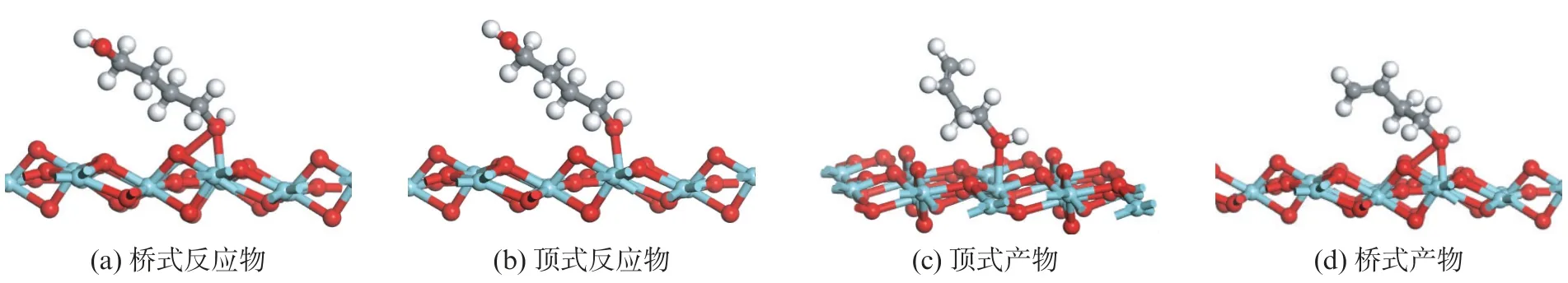
图5 1,4-丁二醇和3-丁烯-1-醇分子在单斜氧化锆(11)表面的单齿吸附构型Fig.5 Monodentate configurations of 1,4-butanediol and 3-buten-1-ol on m -ZrO2(11)surface
反应物(BDO)或产物(BTO)的羟基可以采用桥式或顶式两种方式吸附在氧化物表面[11],表3的计算结果表明,不形成氢键条件下,氧化锆表面无论是反应物,还是产物,桥式和顶式吸附构型的吸附能区别很小。红外漫反射的研究数据表明[3],在高温条件下,更容易形成顶式吸附构型。此外,在氧化锆表面,反应物(BDO)的吸附能要低于产物(BTO)的吸附能,即产物在催化剂表面更加不稳定,更加容易脱附到气相中。这可以解释1,4-丁二醇分子在氧化锆表面只能发生选择性的脱水,而不会发生连续脱水生成丁二烯产物。
表3 在单斜氧化锆(11)表面不同位置单齿吸附1,4-丁二醇分子的键长与吸附能Tab.3 Bond length and adsorption energy of monodentate configurations of 1,4-butanediol on m-ZrO2(11)surface
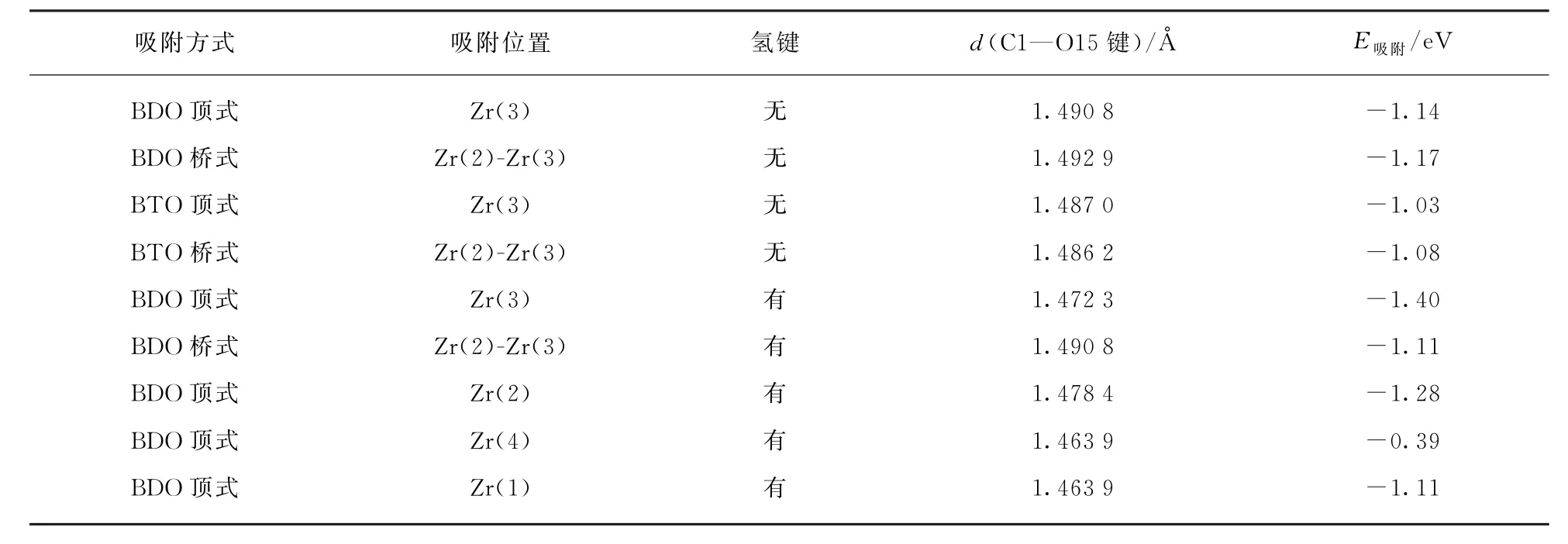
表3 在单斜氧化锆(11)表面不同位置单齿吸附1,4-丁二醇分子的键长与吸附能Tab.3 Bond length and adsorption energy of monodentate configurations of 1,4-butanediol on m-ZrO2(11)surface
表3给出了1,4-丁二醇在氧化锆表面不同Zr位置的吸附能和键长数据。结合福井函数指标分析,Zr(3)是反应性最高的表面锆原子,1,4-丁二醇分子以顶式吸附在Zr(3)位时,释放出最多的能量,最稳定,即Zr(3)是最容易发生反应物分子吸附的位点。而与Zr(1)、Zr(2)、Zr(3)吸附位相比,1,4-丁二醇分子吸附在Zr(4)上时,体系释放出的能量较少,吸附能增大明显。Zr(4)位置相对惰性,很难发生反应物BDO 分子的吸附与活化。表面锆原子与1,4-丁二醇分子发生作用的强度按Zr(3)>Zr(2)>Zr(1)>>Zr(4)的顺序递减。
气相1,4-丁二醇的C—O 键键长分别为1.474 1和1.475 2Å,在催化剂表面形成不同形式的吸附态后,C1-O15键都有一定程度的拉长,碳氧键有所削弱。吸附分子吸附在在氧化物表面后,羟基上的氢原子可以与相邻的氧原子形成氢键,形成氢键后对体系能量略有降低,如上表Zr(3)位吸附的1,4-丁二醇分子。
1,4-丁二醇分子存在两个端位的羟基,除了可以采用单齿形式吸附,也可以采用双齿形式吸附。双齿吸附构型中,1,4-丁二醇分子的两个羟基可以分别与两个相邻的表面锆原子结合,形成站立式的吸附构型(立式);如果1,4-丁二醇分子的两个羟基分别与两个不相邻的表面锆原子结合,则会形成1,4-丁二醇分子结构中碳、氢原子与氧化锆表面更为接近的卧倒式的吸附构型(卧式),典型吸附形式如图6所示。
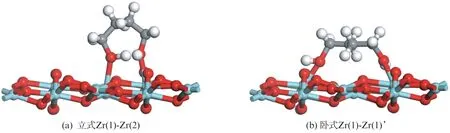
图6 1,4-丁二醇分子在单斜氧化锆(11)表面的双齿吸附构型Fig.6 Bidentate configurations of 1,4-butanediol on m -ZrO2(11)surface
比较表3和表4中的数据发现,采用双齿吸附构型的释放出的吸附能要明显高于单齿吸附构型,反应物分子更容易以双齿构型吸附在催化剂表面。反应物与产物的吸附强度有如下顺序: 双齿吸附反应物分子>单齿吸附反应物分子>单齿吸附产物分子。
表4 在单斜氧化锆(11)表面不同位置双齿吸附构型的BDO分子的键长与吸附能Tab.4 Bond length and adsorption energy of bidentate configurations of 1,4-butanediol on m-ZrO2(11)surface
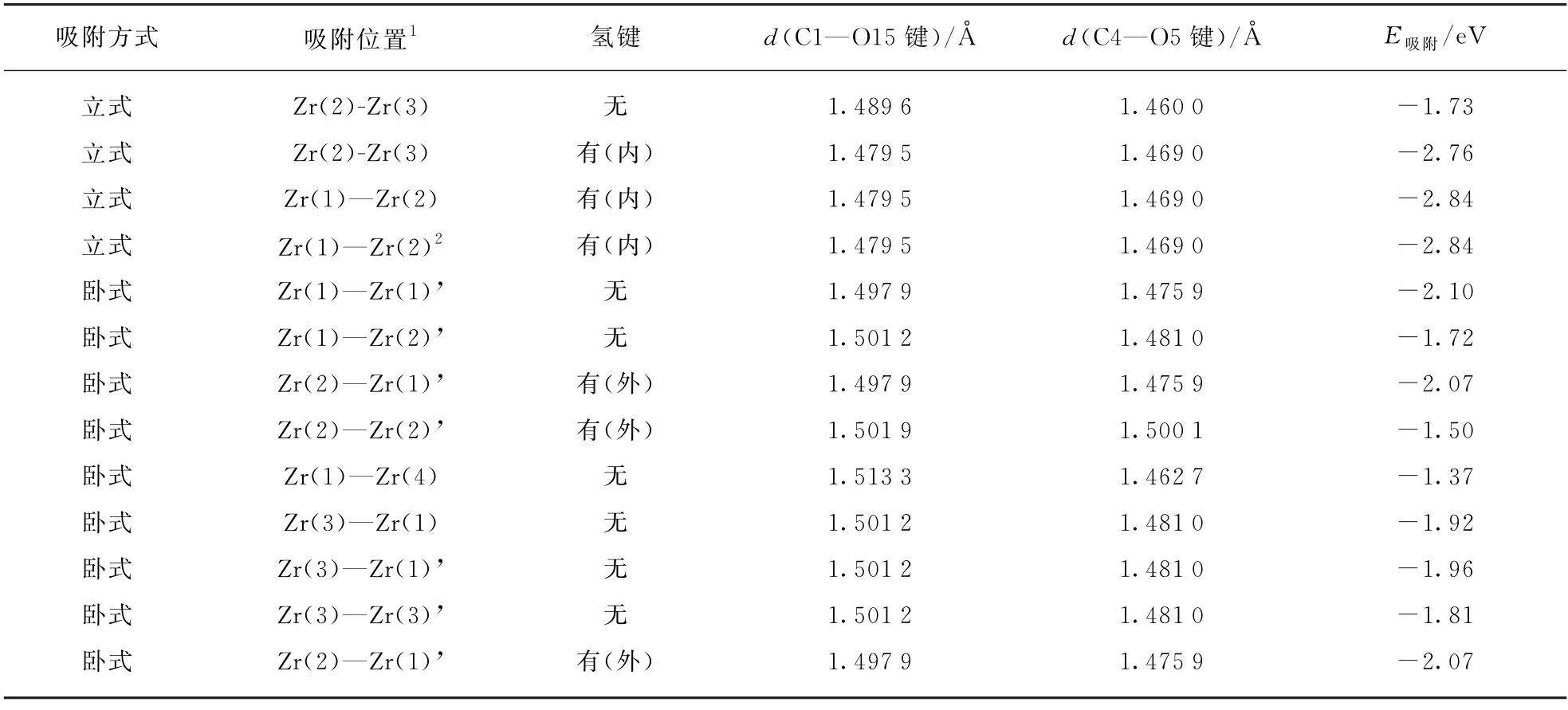
表4 在单斜氧化锆(11)表面不同位置双齿吸附构型的BDO分子的键长与吸附能Tab.4 Bond length and adsorption energy of bidentate configurations of 1,4-butanediol on m-ZrO2(11)surface
注: 1包括相邻周期性位点,2 氢键解离。
上述计算结果也进一步说明了,在大量反应物分子存在条件下,弱吸附在催化剂表面上的丁烯醇产物很容易被1,4-丁二醇反应物替代,脱附进入气相,不会发生进一步脱水反应,生成丁二烯产物。
采用双齿构型吸附在氧化物表面不同位点上的反应物分子,可以采用立式和卧式两种不同的构型。其中,立式构型的反应物形成分子内氢键后的吸附能普遍高于卧式反应物的吸附能。1,4-丁二醇分子两个端位羟基分别锚定在Zr(1)和Zr(2)位的吸附构型具有最低的吸附能;尽管吸附分子羟基上的氢不能与临近的氧原子形成氢键,但是羟基分别锚定在Zr(1)和Zr(1)’位置的吸附分子是依然最稳定的卧式吸附构型。吸附分子羟基上的氢和相邻氧原子发生作用并发生解离不会对吸附能产生影响。
计算Zr(2)-Zr(1)’吸附构型的β-H 与表面氧作用时的吸附能,二者的吸附能几乎没有变化,计算结果说明反应物分子与氧化物表面不出现第3个吸附位点,即不会出现三齿吸附构型。
2.3 吸附分子性能
通过电荷密度差分与布居(Mulliken)分析来跟踪1,4-丁二醇分子形成不同吸附构型后的电子密度和相关原子得失电子的状态。图7给出了1,4-丁二醇分子在氧化物表面形成不同双齿吸附构型后的电荷密度差分图。图中可以观察到,反应物分子通过两个羟基在氧化锆表面形成立式和卧式两种双齿吸附构型后,体系电子云密度变化最大的位置都出现在C—O 键和O—Zr键的位置。
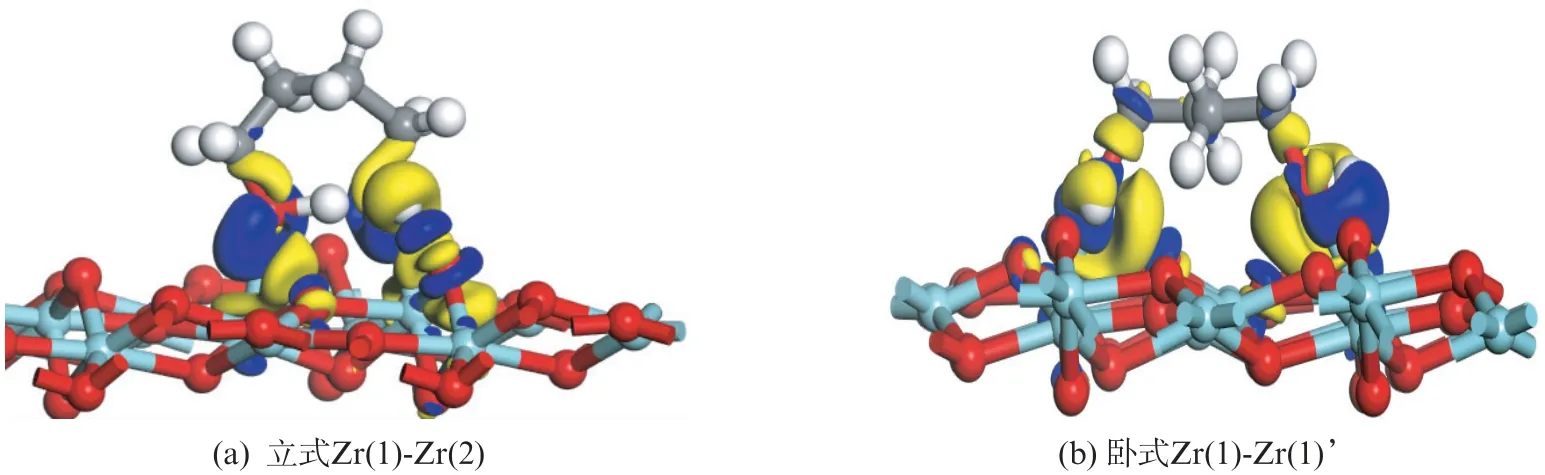
图7 1,4-丁二醇分子在氧化锆(11)表面双齿吸附构型的电荷密度差分Fig.7 Charge density difference for bidentate configurations of 1,4-butanediol on m -ZrO2(11)surface
无论是立式还是卧式的吸附构型,C—O 键区域的电子密度减小(黄色区域),而O—Zr键区域电子密度增大(蓝色区域),吸附在氧化锆表面后,两个C—O 键强度都被削弱了。此外,立式吸附构型中,没有形成氢键的羟基氢与氧化锆表面的二配位氧距离接近,也发生了明显的电子偏移,这个氢原子很容易发生解离,并与表面二配位氧形成表面羟基。
1,4-丁二醇吸附前后羟基与锆原子的布居(Mulliken)分析如表5所示。在立式的吸附构型中,两个锚定位置的电子转移的趋势相同,氧原子是接受电子中心,价态呈下降的趋势;而H 和Zr原子都是给电子中心,价态上升。而在卧式的吸附构型中,锚定位置的电子转移方向与前者有明显区别。Zr(1)—O(15)—H(16)锚定位与立式的变化趋势相同;而Zr(1)’—O(5)—H(14)锚定位呈现出相反的趋势,Zr原子接受电子,价态下降,氧原子成为弱的供电子中心,价态上升。由此可见,1,4-丁二醇在氧化锆表面形成不同的双齿吸附构型,不仅具有不同的稳定性(表4吸附能数据),而且也可能存在不同的电子转移机制。
表5 1,4-丁二醇吸附前后羟基与锆原子的布居(Mulliken)分析Tab.5 Mulliken population analysis of Zr atoms and OH groups before and after 1,4-butanediol adsorption on m -ZrO211)surface
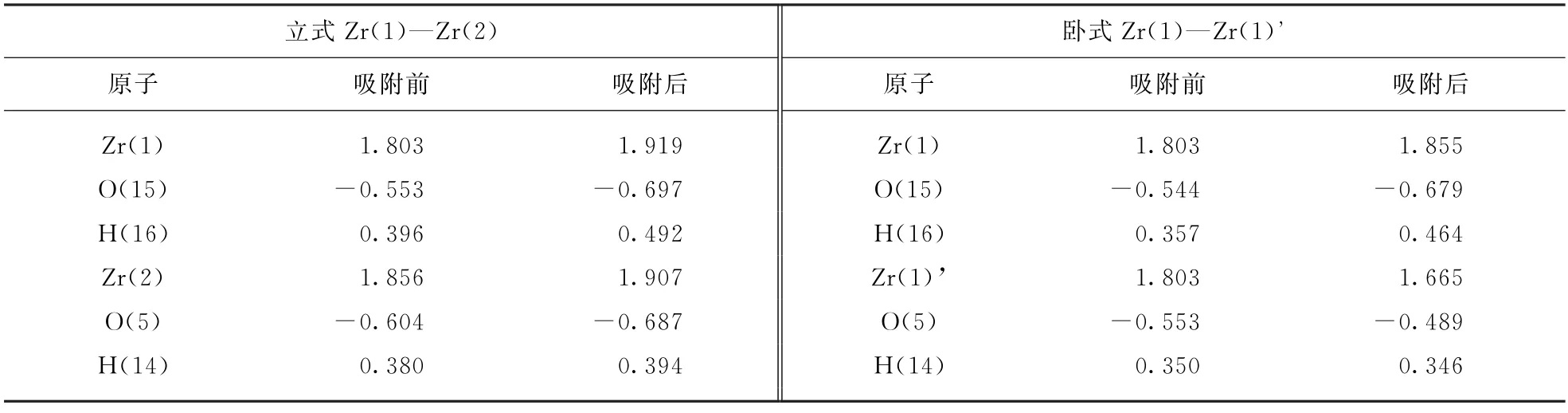
表5 1,4-丁二醇吸附前后羟基与锆原子的布居(Mulliken)分析Tab.5 Mulliken population analysis of Zr atoms and OH groups before and after 1,4-butanediol adsorption on m -ZrO211)surface
2.4 β-H 脱除基元反应步骤过渡态
在氧化锆表面吸附活化的1,4-丁二醇反应物,脱去一个β-H 得到目标产物丁烯醇是合理的基元反应步骤[9,20]。图8给出了立式反应物与卧式反应物在(11)面上β-H 脱除基元反应步骤过渡态。两种吸附构型在锆氧化物(11)面都可以实现β-H的脱除。
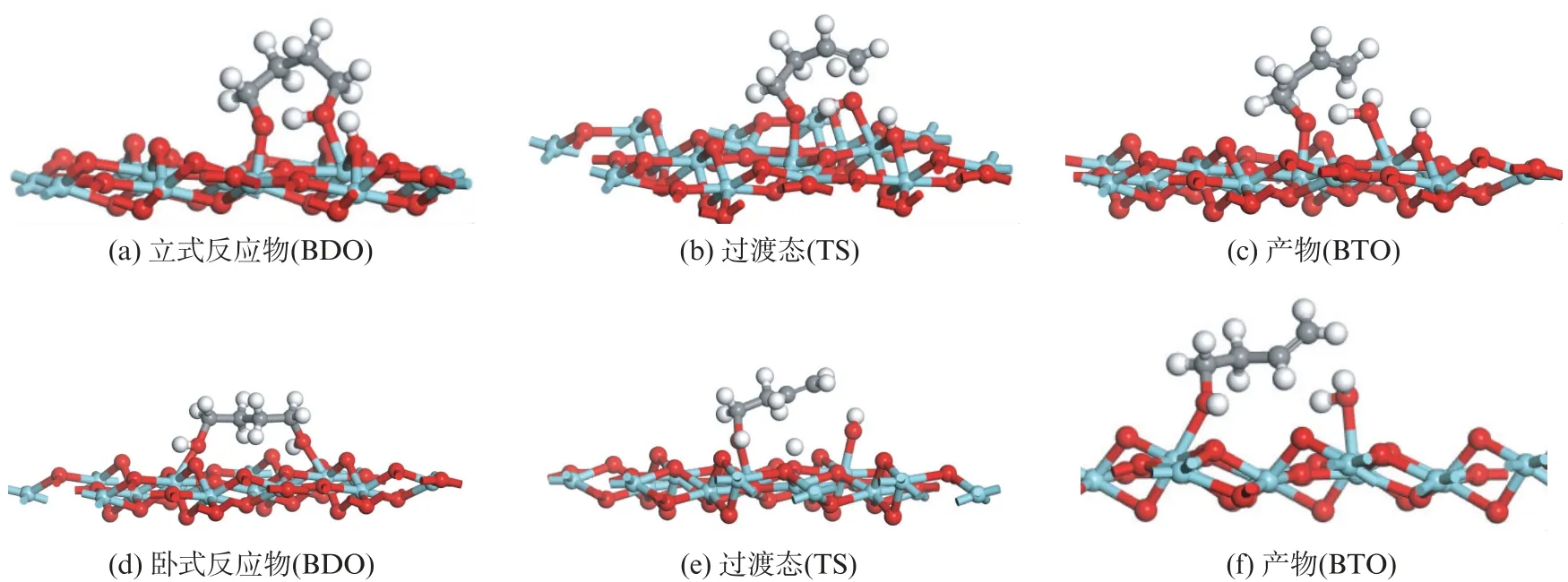
图8 表面1,4-丁二醇β-H 脱除基元反应步骤过渡态Fig.8 Transition states forβ-H removal elementary reaction of adsorbed 1,4-butanediol
立式构型丁二醇反应物的羟基氢原子首先发生解离,与表面二配位氧形成表面羟基,同时分子内氢键的氢原子转移到发生解离的氧原子上,β-H 脱落的同时碳氧键断裂,醇分子形成一个双键,生成单齿吸附的产物分子。如前分析,生成的产物分子在反应条件下可以快速脱附到气相中。卧式的1,4-丁二醇吸附构型,可以发生类似的过程脱除β-H。然而,卧式吸附分子脱除β-H 的能垒仅为1.33 e V,远小于立式的吸附构型的2.97 e V,因此,尽管立式的吸附能更低,但是卧式的吸附构型更加有利于β-H 的脱除,实际的选择性脱水过程倾向于按照卧式1,4-丁二醇吸附构型进行。
3 结论
采用第一性原理计算方法以及周期性超胞模型研究了1,4-丁二醇在单斜二氧化锆(m-ZrO2)的(11)面的吸附。研究表明,反应物分子通过两个羟基与单斜氧化锆(11)面配位不饱和的锆原子结合,采用双齿吸附构型吸附在氧化锆表面。单齿吸附的反应物以及丁烯醇产物在氧化锆表面的吸附不能稳定存在。
氧化锆表面不同位点的双齿吸附构型中,立式和卧式是两种稳定的吸附构型。吸附在氧化物表面后,反应物分子的C—O 键的电子部分转移到O—Zr键的位置,C—O 键强度被削弱、活化,有利于C—O键断裂发生脱水反应。1,4-丁二醇在氧化锆表面形成的双齿吸附构型,两个羟基锚定位置与表面锆原子的结合强度不同,而且存在不同的电子转移机制。
卧式的吸附构型更加有利于β-H 的脱除,实际的选择性脱水过程倾向于按照卧式吸附构型脱去β-H的基元反应步骤进行。
