多相催化中自由基及其反应的研究新进展
2023-01-31徐冰君
田 昊 ,徐冰君
(1.北京大学 化学与分子工程学院,北京 100871;2.北京分子科学国家研究中心,北京 100871)
多相催化过程可以降低反应温度和提升产物选择性,许多与人类社会发展密切相关的化工生产过程,例如合成氨、石油炼制以及合成气转化,都是在固体催化剂的帮助下实现的。多相催化的传统模型将气态或液态的反应物在固体催化剂表面进行反应的历程分为5个步骤:1) 反应物从流体中扩散至催化剂表面附近(包含外扩散和内扩散);2) 反应物在催化剂表面吸附;3) 反应物在催化剂表面反应,转化为产物;4) 产物从催化剂表面脱附;5) 产物从催化剂表面附近扩散至流体中[1]。其中的核心步骤是第2和3步,即反应物在催化剂表面的吸附和反应。对于最常见的双分子反应A+B,根据A 和B在催化剂表面的吸附及反应性质的不同,可以根据经典的动力学模型进行描述(图1): 当两种反应物分子均在催化剂表面吸附,而后吸附的物种发生反应时,采用Langmuir-Hinshelwood模型描述催化反应动力学;当反应物之一发生吸附,另一种反应物不发生吸附直接与前者反应时,分析催化动力学则采用Eley-Rideal模型[1]。

图1 多相催化双分子反应的经典“吸附-反应”动力学模型和生成自由基的动力学模型的示意图Fig.1 Schematic illustration of the classical“ adsorptionreaction”kinetic model and the kinetic model of free radical formation in bimolecular reaction in heterogeneous catalysis
然而,在多相催化中,反应物在催化位点转化为产物一般包含多个基元反应,此时在反应物与产物中间存在若干能量相对较低的反应中间体,当反应中间体在催化剂表面吸附较弱时,会发生脱附进入流体相中。如果这些脱附的反应中间体中含有不成对的电子,则相当于多相催化过程产生了自由基。自由基物种反应性质活泼,可以不依赖固体催化剂的活性位点直接与反应物发生反应,从而使反应动力学偏离经典的“吸附-反应”动力学模型(图1)。上述反应中间体在催化剂表面发生脱附并产生自由基的情形常见于烷烃作为反应物或产物的反应中。这是因为烷烃分子是饱和的碳氢化合物,其中C—H 单键以及C—C单键的键能都较高,一般很难发生解离吸附,同时碳和氢的电负性相当,C—H 键的极性很小,无论是C—H 断键还是C—C断键都倾向于发生均裂,生成两个含有不成对电子基团或原子。因此,若C—H 键活化反应的中间体发生脱附,通常会产生自由基物种。此外,自由基聚合是碳链增长的重要反应机理,多相催化中许多碳链增长过程都与自由基有关[2-3]。综上所述,本文将以多相催化中的两类重要反应类型,即碳氢键活化反应与碳碳键偶联反应,作为讨论对象,首先回顾多相催化中自由基反应研究的历史,并介绍研究多相催化中自由基反应的实验研究手段,然后总结目前研究中关于自由基反应对多相催化过程影响的认识,最后对这一领域未来的研究方向进行简单的展望。需要指出的是,本文的讨论主要是集中在传统的多相热催化领域,在近几年兴起的多相光催化、电催化体系中,也有很多关于自由基反应贡献的研究与讨论[4-8],本文的主要内容并不涉及这些领域。
1 多相催化中的自由基反应
1.1 早期的研究和认识
氧气分子是一种稳定的双自由基(·O =O·),O2作为反应物的许多非催化反应都是通过自由基机理进行的,例如燃烧。多相催化中早期关于气相自由基在多相催化中的贡献同样是在O2作为反应物的选择性氧化反应中观察到的。Keulks及其团队在钼酸铋催化的丙烯选择性氧化反应中发现,在管式固定床反应器中,催化剂床层后的空体积对丙烯转化率和产物分布具有很大影响,随着催化剂床层后的空体积增大,丙烯转化率逐渐提高,但目标产物丙烯醛的选择性降低,CO 和CO2的选择性升高,同时伴随少量环氧丙烷的生成[9-10]。根据上述实验现象,作者认为丙烯醛的生成反应是在催化剂表面进行的,而生成CO 和CO2的氧化反应则是一个表面引发的气相自由基反应过程[9-10]。在这一过程中,自由基反应导致了副反应的发生,降低了多相催化的选择性。
另一个早期关于多相催化中自由基反应的认识来自于另一个有O2参与的反应,甲烷氧化偶联反应(Oxidative Coupling of Methane,OCM),即CH4在O2存在下反应生成C2H6以及C2H4的过程。降低反应物CH4和O2在催化剂床层的停留时间,可以发现C2H6是甲烷氧化偶联反应的初级产物[11]。Nelson等[12]研究了Li/MgO 催化的CH4和CD4的氧化偶联反应,结果发现乙烷产物只有C2H6、C2D6和CD3CH3这3种,乙烯产物只有C2H4、C2D4和CD2CH2这3种,且几种产物的分布可以根据动力学同位素效应(Kinetic Isotope Effect,KIE)进行计算,初步说明了C2H6是通过气相CH3·自由基的偶联反应生成的,而C2H4是通过C2H6的气相氧化脱氢反应产生的。
在上述两个例子中,研究者们是通过一些间接手段,即改变反应器空体积以及同位素实验,说明了可能存在的自由基反应。此类间接方法不依赖于仪器硬件,在实验设计较为合理的前提下,可以为多相催化中自由基反应的存在提供实验依据。然而,自由基反应存在最直接的证据仍然是自由基的捕获和检测,即直接通过实验表征的手段。下面将介绍多相催化中直接检测自由基物种的实验技术。
1.2 多相催化中检测自由基的实验技术
1.2.1 质谱(Mass Spectrometry,MS)
质谱具有高灵敏度,在采用合适的进样技术时,具备在线检测自由基的能力。Robertson 等[13]在1949年首次报道了应用质谱检测到CH4在1 000℃下,经过Pt表面时产生的CH3·自由基。然而,传统的质谱采用电子碰撞(Electron-Impact,EI)电离技术,其带来的强碎片化现象干扰了相同质荷比的分子与其他分子所产生的碎片之间的区分。
为更好地研究气相自由基物种,逐渐开始有应用其他电离技术的文献报道,特别是真空紫外光电离(Vacuum Ultraviolet Photoionization,VUV-PI)技术与激光共振增强多光子电离(Resonance Enhanced Multiphoton Ionization,REMPI)技术,与质谱联用研究气相自由基的工作[7]。激光共振增强多光子电离技术具有极高的分辨率,在同分异构小分子和芳香化合物大分子的区分上有较好的效果。但由于选择规则的限制,激光共振增强多光子电离技术并不是一种检测气相自由基的通用实验手段,更适用于研究特定的简单体系,而不是多种组成的复杂反应体系[14]。相比之下,真空紫外光电离技术可以采用同步辐射光源,将其与分子束质谱联用(Synchrotron Vacuum Ultraviolet Photoionization Mass Spectroscopy,SVUV-PIMS),可以实现最小化碎片干扰,通过改变光子能量,可以实现自由基物种的鉴定[15]。应用SVUV-PIMS技术进行自由基检测的实验结果将在后面的部分进行介绍。
对于光电离质谱相差比较大的同分异构体,SVUV-PIMS可以实现较好地区分。但当同分异构体之间的光电离质谱非常接近时,SVUV-PIMS技术的分辨率就不足以区分了[14]。为提高分辨率,可以采用成像光电子-光离子复合谱技术(Photoelectron Photoion Coincidence,PEPICO)(图2)[16],即同时记录光电子的动能以及来自于同一电离事件的光离子的延时,获得质量选择阈值光电子谱(mass-selected Threshold Photoelectron Spectra,ms-TPES)[14]。PEPICO 相比于SVUV-PIMS对同分异构体具有更高的分辨率,应用PEPICO 在多相催化中进行自由基检测的实验结果也会在后面的部分进行介绍。
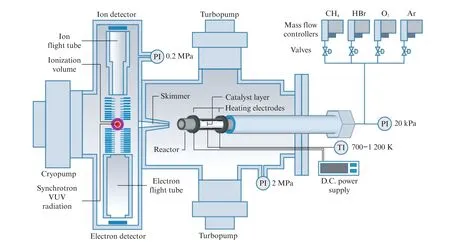
图2 用于多相催化中自由基检测的PEPICO 反应器装置[16]Fig.2 PEPICO reactor for free radical detection in heterogeneous catalysis[16]
1.2.2 其它技术
除质谱技术外,电子顺磁共振(Electron Spin Resonance,ESR)、红外光谱、激光激发的荧光光谱等技术在结构合理的检测池辅助下也可以提供自由基结构的直接信息[17]。例如,Lunsford课题组应用基质隔离的电子顺磁共振(Matrix Isolation Electron Spin Resonance,MIESR)技术在Li/Mg O 等一系列氧化物催化的甲烷氧化偶联反应中检测到了气相中的CH3·自由基[11]。但是需要指出的是,上述表征技术运用到多相催化中的自由基检测通常需要降低反应体系的压力,进而可能使实验结果偏离真实条件,并且一般只能检测出浓度较高的自由基物种。
2 多相催化碳氢键活化中的自由基反应
2.1 烷烃氧化脱氢制烯烃
烷烃氧化脱氢生成烯烃反应的速控步骤一般为烷烃第一个C—H 键的断裂。以丙烷为例,烷烃氧化脱氢的反应方程式为C3H8+O2C3H6+H2O。
烷烃氧化脱氢反应与烷烃完全燃烧反应的机理类似,而后者的反应机理已被证实为自由基链反应[18]。受此启发,研究者们尝试在丙烷氧化脱氢反应中引入自由基链反应,以提升其效率。Annamalai等[19]在丙烷氧化脱氢中引入了少量NO,对比了气相NO 和V2O5固体催化剂对丙烷氧化脱氢反应产物分布和机理的影响。实验结果显示,NO 分压与C3H8转化率成正比,以NO 作为催化剂产物C3H6的选择性在80%左右,C2H4是最主要的副产物,不同于以CO 和CO2为主要副产物的V2O5催化剂。研究者认为NO 在体系中以NO—NO2—HONO 的自由基链反应的形式产生具有强夺氢活性的OH·气相自由基,从而驱动C3H8转化。厦门大学王野课题组的研究发现,在HCl存在的条件下,CeO2对丙烷氧化脱氢反应具有良好的催化性能[20]。引入少量NiO 对CeO2的性质进行修饰,可以在773 K 下实现69%的丙烷转化率以及80%的丙烯选择性,丙烯的单程收率为55%。在没有HCl存在的条件下,CO2为反应的主产物,其选择性高达93%,这说明HCl在丙烷氧化脱氢制丙烯的过程中发挥了不可或缺的作用[20]。结合在拉曼光谱中观察到的过氧化物物种),作者提出在CeO2表面氧缺位吸附的氧气活化了HCl并生成了Cl·自由基物种,生成的Cl·自由基物种进而与C3H8反应活化C—H 键,并生成C3H6作为主产物[20]。
另一个在烷烃氧化脱氢反应中与自由基链反应存在显著关联的例子是h-BN。2016年h-BN 被报道具有烷烃氧化脱氢活性[21-22],Hermans课题组的后续研究表明,h-BN 催化的烷烃氧化脱氢中气相反应对其活性有较大贡献。首先,h-BN 催化烷烃氧化脱氢的产物分布与前文中NO 的结果类似,明显不同于V2O5[23]。其次,固定催化剂h-BN 的质量,仅改变稀释剂SiC和催化剂质量的比例,发现h-BN 的催化活性随床层体积的增大而增大[24]。对此,Hermans课题组提出了一套自由基链反应机理对h-BN 的催化性质进行解释[25]。气相自由基链反应的观点也受到了气相燃烧自由基理论计算的支持[26]。徐冰君课题组以乙烷和丙烷的混合烷烃作为反应物,研究h-BN 催化的氧化脱氢过程[27]。结果表明,丙烷的存在可以显著促进乙烷脱氢转化的速率,在580℃下,少量丙烷的引入可以使乙烷的转化率提高47%(图3(a))。同时,固定乙烷分压不变时,乙烷氧化脱氢速率对丙烷分压的表观反应级数为1(图3(b))。这说明烷烃表观的二级级数可以解耦为自由基引发剂和反应物两种角色。选取环氧丙烷作为指示分子,采用程序升温氧化的实验方法对比丙烷和丙烯的性质,发现丙烷生成的环氧丙烷约为丙烯的5倍,说明由丙烷引发生成的活性物种是h-BN 体系中主要的夺氢氧化活性物种。基于上述实验结果,研究者提出了基于气相自由基链反应的h-BN 氧化脱氢机理,并采用稳态近似推导了烷烃转化速率方程。由速率方程拟合得到的动力学参数能够很好地匹配乙烷丙烷共氧化脱氢的实验结果(图3(c))[27]。
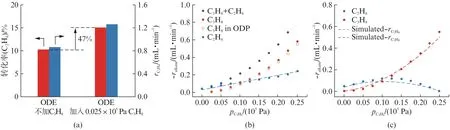
图3 h-BN 催化的乙烷丙烷氧化共脱氢过程[27]Fig.3 Oxidative co-dehydrogenation of ethane and propane catalyzed by h-BN[27]
尽管自由基链反应可以解释h-BN 以及大多数硼基催化剂在烷烃氧化脱氢中的实验结果,但需要指出的是,目前这一理论仍存在许多不足,其中之一是缺少证明自由基存在的直接实验证据。Zhang等[28]采用SVUV-PIMS试图原位检测h-BN 催化烷烃氧化脱氢中的活性自由基,然而仅检测到了CH3·自由基,并没有观察到与硼物种或活性氧物种有关的自由基。这一领域与自由基反应的关联仍有待进一步研究。
2.2 甲烷的官能化反应
甲烷中C—H 键的键能高达439.3 kJ·mol-1,甲烷的选择性转化被称为催化领域的“圣杯”。利用自由基活泼的反应性,可以活化CH4中的C—H 键。Javier Pérez-Ramírez课题组研究了(VO)2P2O7和Eu OBr催化的甲烷氧溴化反应,应用PEPICO 光谱在反应中直接检测到了Br·和CH3·自由基物种[16]。结合动力学分析以及DFT 的计算结果,研究者认为甲烷催化氧溴化反应主要由表面催化的HBr氧化反应和气相中的CH4溴化反应组成。与非催化的甲烷溴化反应类似,甲烷与Br·自由基在气相中的碳氢断键反应是速控步骤。催化剂表面在这一过程中起到了再生HBr生成Br·和Br2的作用,从而抑制了自由基链反应中的链终止反应[16]。Ferdi Schüth 课题组发现以三氯异氰尿酸(Trichlroisocyanuric Acid,TCCA)作为氯化试剂,在Lewis酸的催化下,可以使CH4高效地转化为CH3Cl[29]。在机械力摇动的辅助下,CH4和TCCA的反应温度可以从200℃下降到90℃,CH3Cl的选择性从66%提升为95%。作者提出了一套自由基链反应机理解释TCCA的高活性和选择性[29]。中科院大连化学物理所的包信和课题组在Fe催化的甲烷非氧化转化的反应中引入氢的供体,即1,2,3,4-四氢萘(1,2,3,4-tetrahydronaphthalene,THN)和苯,提升了CH4的转化速率[30]。同位素实验的结果表明,CH4转化速率的提升是通过与H·自由基的均相反应实现的:H·+CH4H2+CH3·[30]。
3 多相催化碳碳键偶联中的自由基反应
3.1 甲烷的氧化偶联反应
甲烷氧化偶联的催化机理目前已在多个催化体系中被证实是一个多相和均相反应共同存在的过程:CH4首先在催化剂表面的活性位点上活化生成气相甲基自由基CH3·,随后CH3·在气相中发生均相的自由基偶联反应生成C2H6,一部分C2H6会继续发生脱氢反应生成C2H4[11,31]。近几年随着表征技术以及材料合成技术的发展,这一领域不断有新的发现。中国科学技术大学黄伟新课题组利用SVUV-PIMS鉴别了甲烷氧化偶联以及乙烷氧化脱氢反应中气相中间物种[32]。在Li/MgO 催化剂的体系中,观察到了CH2·、CH3·、C2H5·、CH3OO·、C2H5OO·、CH3OOH 以及C2H5OOH 等中间物种。基于上述物种的发现,研究者建立了甲烷氧化偶联以及乙烷氧化脱氢的反应网络[32]。在此基础上,利用SVUV-PIMS研究了Li-MgO 在甲烷氧化偶联反应中的构效关系[33]。结果显示,Li的加入使Mg O 发生了重构,MgO{110}面四配位的Mg2+位点数量增加。Mg O{110}面四配位的Mg2+位点可以活化CH4生成CH3·自由基,并能避免甲基自由基的深度脱氢以及氧化的副反应[33]。Han等[34]将铁橄榄石和石英通过熔融的方法将铁位点高度分散地限域在方英石相(cristobalite)中合成了Fe@CRS催化剂。Fe@CRS催化剂在甲烷制烯烃、芳烃及氢气的反应中实现了100 h的稳定运行,甲烷转化率为5.8%~6.9%,C2选择性为86.2%。计算结果表明,晶格限域的Fe位点相比于Fe3C更有利于CH3·自由基的生成及脱附,研究者认为C2等多碳产物是通过CH3·自由基在气相中的偶联反应生成的[27]。
3.2 甲醇制烯烃反应的碳链增长过程
Javier Pérez-Ramírez课题组应用PEPICO 光谱对分子筛催化的甲醇和一氯甲烷制碳氢化合物的过程在反应条件下进行了原位动态表征,以鉴别反应中间体物种[35]。结果显示,甲基自由基物种同时存在于CH3OH 和CH3Cl转化的反应路径中,而烯酮中间体(C2H2O)只存在于CH3OH 转化生成碳氢化合物的路径中。研究者认为,CH3·自由基和C2H2O 两种中间体对应着两种碳链增长机理: 一种是通过CH3·自由基的甲基化反应实现碳链增长过程,这一机理是CH3Cl生成长链碳氢化合物的最主要机理;另一种是有C2H2O 重组反应参与的碳链增长机理,包含羟醛缩合、脱水以及含氧物种与CH3·自由基的甲基化反应等[35]。
4 总结与展望
在多相催化的研究中,由于经典的“吸附-反应”式模型带来的影响,自由基反应的贡献往往被忽略。如本文所述,在一些碳氢键活化以及碳碳键偶联的反应中,体系中自由基反应对催化性质有较大影响,有时甚至主导了反应过程,固体催化剂在其中扮演自由基链反应引发剂的角色。多相催化中自由基反应的性质仍有待更加深入和广泛的研究。在研究手段上,SVUV-PIMS、PEPICO 等技术已经在直接检测自由基方面发挥了巨大作用,然而这些技术的检测限和分辨率对于某些复杂体系、低浓度自由基仍存在不足,继续发展能够在真实反应条件下检测或捕捉自由基的实验技术将是推动这一领域取得实质进展的基础和关键。
