狼疮性肾炎潜在蛋白质生物标志物的生物信息学分析
2023-01-30刘南池王雁飞周燕魏玉娇秦双马瑞霞
刘南池,王雁飞,周燕,魏玉娇,秦双,马瑞霞
(青岛大学附属医院肾病科,山东 青岛 266000)
狼疮性肾炎(LN)是系统性红斑狼疮(SLE)病人常见且严重的并发症,也是导致肾衰竭和病人死亡的重要原因[1]。LN的发病除了与免疫混合物的形成、细胞因子异常有关以外,还具有明显的遗传相关性。然而,迄今确定的众多与LN相关基因只解释了20%的疾病遗传性[2]。缺失的遗传性可能来自基因和环境(病毒感染、药物作用等)之间复杂的相互作用,以及基因表达的调控被破坏等[3]。因此,探究LN相关的遗传标志物对疾病的早期诊断和预后评估有重要意义。随着生物信息学分析和基因芯片技术的发展,整个转录组的基因表达谱分析越来越多地被用于探索致病相关基因、对不同类型的疾病进行分类和预测临床转归[4]。然而,目前对SLE和LN的生物信息学分析研究还很少。本研究选取GSE32591的数据集,确定LN病人中差异表达基因(DEGs),并对DEGs进行基因本体(GO)分析和KEGG分析,使用Cytoscape软件构建DEGs的蛋白质相互作用(PPI)网络,预测其中的核心基因及发挥调控作用的转录因子,为探讨LN的发生发展提供依据。
1 材料与方法
1.1 数据采集
以“Lupus Nephritis”为关键词,在基因表达综合(GEO)数据库[5](http://www.ncbi.nlm.nih.gov/geo/)中搜索数据集,筛选标准:①芯片研究类型为表达谱芯片;②无其他干预措施;③样本分组为正常组和疾病组;④提供基因符号(Gene symbol)的注释信息。最终选取了编号为GSE32591的项目,其数据为通过微阵列分析的微分解的肾脏活检组织(来源于美国密西根大学)的转录组,共包括93例样本。其中取自肾小管间质组织的LN病人样本32例,正常对照组样本15例;取自肾小球组织的LN病人样本32例,正常对照组样本14例。该数据集微阵列分析是基于GPL14663 Affymetrix GeneChip Human Genome HG-U133A Custom CDF平台进行的。
1.2 DEGs确定
通过GEO2R程序[6]分别确定LN样本与正常样本肾小管间质和肾小球组织中的DEGs。筛选标准设定为矫正后P值<0.05,|logFold Change|≥1。然后应用Venn图将肾小球DEGs和肾小管间质DEGs取交集。
1.3 功能和通路富集分析
使用DAVID[7](http://david.ncifcrf.gov/)对肾小球组织DEGs和肾小管间质DEGs交集部分的DEGs进行GO分析和KEGG分析,明确基因富集的通路和功能,设置阈值为P<0.05,错误发现率(FDR)<0.05。GO[8]分析按照生物途径、分子功能和细胞定位对基因进行注释分类,KEGG[9]分析旨在通过代谢通路的分析诠释基因功能。
1.4 PPI网络的构建
使用互作基因数据库的检索工具STRING[10](https://string-db.org/cgi/)构建相互作用基因的网络,输入多个基因、蛋白质名称,得到总结了特定蛋白质组预测关联的网络视图。通过Cytoscape软件将PPI网络可视化,隐藏无相关联系基因,得到合理的布局。
1.5 网络分析
在PPI网络中,使用Cytoscape的cytohubba插件预测核心基因[11],结合12种算法的结果合并分析,选出其中出现频数最高的基因确定为核心基因。使用Cytoscape软件的MCODE[12]插件在庞大的蛋白网络中进行基因功能模块聚类构建,确定高度连接的基因集。这些高度互连的区域被称为子网络。本研究设定影响子网络大小的节点评分截断值(NSC)=0.2,K-Core=2[13]。使用Cytoscape软件的iRegulon插件预测子网络的转录因子[14],所选的转录因子信息来自SwissRegon、Jaspar、Encode、Transfac和Hmer数据库[15]。在分析结果中选取归一化富集分数(NES)>10的转录因子,并绘制网络图。
1.6 核心基因差异表达分析
在Nephroseq[16](http://www.nephroseq.org)中依次检索确定的核心基因,设定LN为筛选条件,选择相关研究,进行Meta分析综合比较。
2 结 果
2.1 LN相关DEGs
在GSE32591的数据中,通过GEO2R确定了LN病人和正常对照组之间的DEGs,在肾小管间质样本中共确定了130个DEGs,其中有25个下调基因,105个上调基因;在肾小球样本中共确定了352个DEGs,其中102个下调基因,250个上调基因。将肾小球和肾小管间质的DEGs进行比较,使用Venn图取交集,确定了66个共有的DEGs(图1),其中11个下调基因,55个上调基因。

G:肾小球样本的DEGs;T:肾小管间质样本的DEGs。图1 DEGs的Venn图
2.2 功能和通路富集分析
GO分析显示,在生物学过程层面,基因显著富集于防御病毒、对病毒的反应、Ⅰ型干扰素(IFN)信号通路、先天免疫反应、病毒基因组复制的负调控、干扰素-γ介导的信号通路、对干扰素-β的反应、对干扰素-α的反应8个方面,而在细胞组分、分子功能层面未发现显著富集。KEGG分析显示,有9个基因参与甲型流感,8个基因参与单纯疱疹病毒感染。见表1。

表1 GO和KEGG分析结果
2.3 PPI网络
使用STRING构建PPI并通过Cytoscape软件可视化,在网络中,循环节点代表基因,两个节点之间的连线代表基因之间的相互作用(图2)。此网络包括60个基因点和432条相互作用线,其中52个上调基因,8个下调基因。可见,在确定的66个DEGs中有6个基因未参与构成网络;并且在LN病人DEGs中,以上调表达的基因为主。

红色为上调基因,共52个;蓝色为下调基因,共8个。图2 PPI网络
2.4 网络分析
2.4.1核心基因 应用cytohubba的12种算法最终预测2’-5’-寡腺苷酸合成酶1(OAS1)、IFIT3、三联基序22(TRIM22)、IFIT1(频数分别为8、7、7、6)为核心基因。见表2。在MCODE插件的分析结果中,选定了评分最高的一组基因群集(MCODE评分为24.72)作为本次的研究对象,共包括26个基因、309条相互作用关系连线。在此功能模块中,26个基因全部为表达上调的基因,且高度相互连接,在网络中起到了重要作用(图3)。其中,作用最强的基因为TRIM22、磷脂加扰酶1(PLSCR1)。
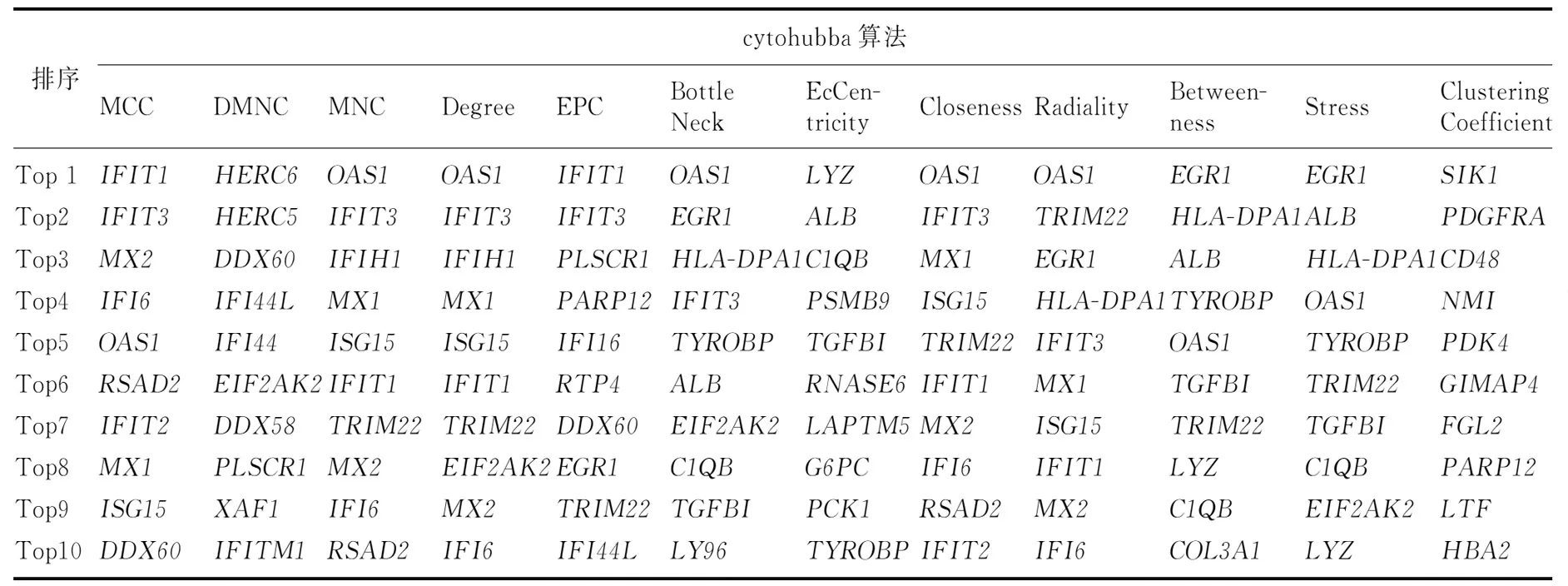
表2 cytohubba的12种算法及结果

圆形:基因,面积大小代表作用程度强弱;蓝色:作用较强的基因;橙色:作用较弱的基因。图3 核心基因子网络
2.4.2转录因子 用Cytoscape 的iRegulon插件对子网络中26个关键基因的转录因子进行分析,结果显示,NES>10的因子分别为信号传导及转录激活因子2(STAT2)(NES=17.482,靶向=25)和STAT1(NES=16.109,靶向=23)。二者对子网络中的大部分基因起直接调控作用(图4)。
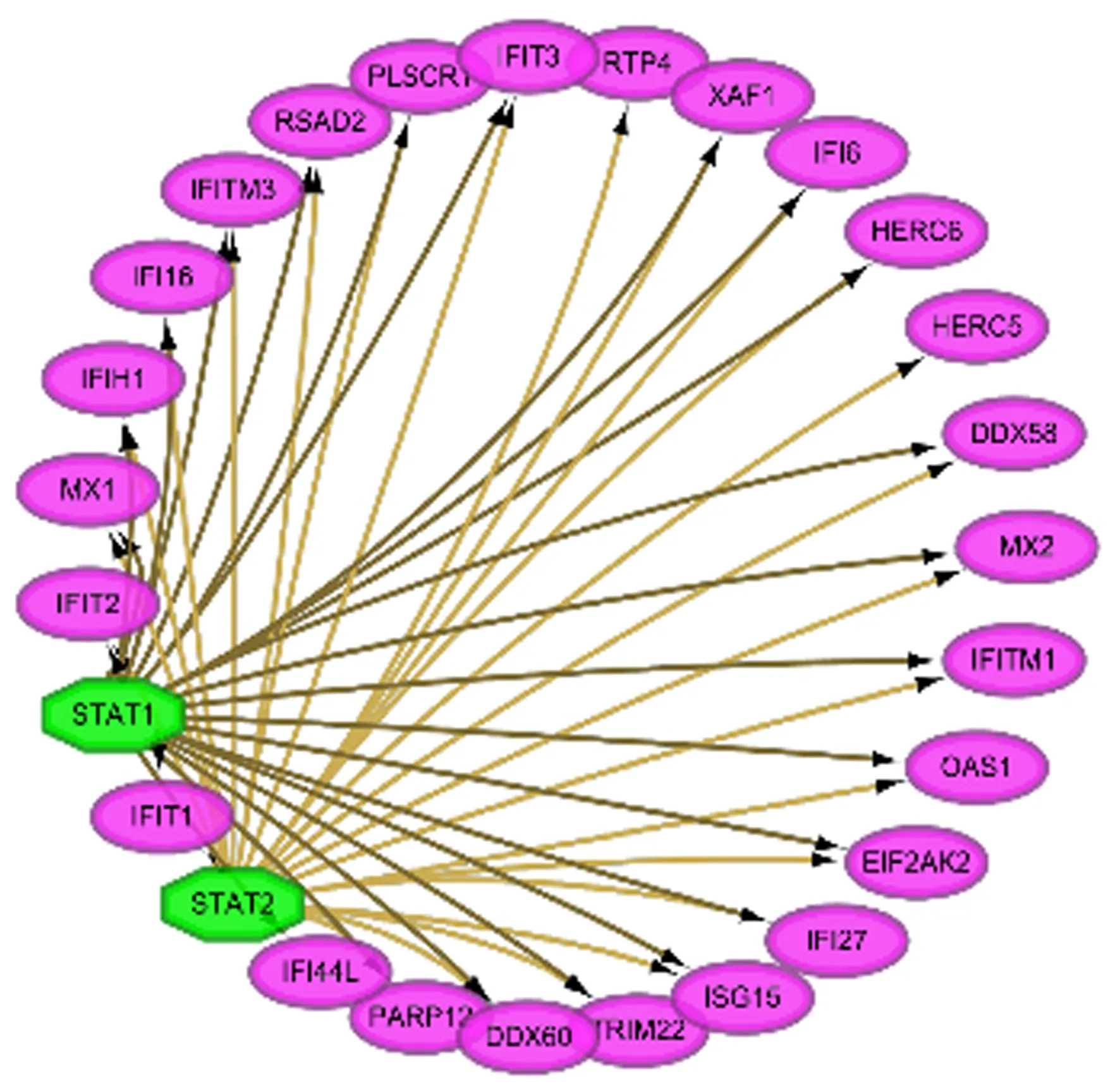
紫色表示基因,绿色表示转录因子。图4 转录因子网络图
2.5 核心基因的差异表达分析
用Nephroseq进一步验证4个核心基因的表达,证实IFIT1、IFIT3、TRIM22和OAS1在LN样本和正常样本中的表达存在显著差异,并且皆表现为过表达。表明IFIT1、IFIT3、TRIM22以及OAS1是LN中重要的上调基因。
3 讨 论
本研究对DEGs的GO分析显示,LN病程与病毒入侵有关,提示病毒免疫与自身免疫具有相关性。在慢性病毒感染期间,CD8+T细胞在抗原暴露和CD4+T细胞缺乏的共刺激下耗尽衰竭,不但使得病毒清除过程受阻碍,同时也影响了自身免疫性疾病的进展[17]。因此,在LN中病毒免疫与自身免疫密不可分。GO分析还显示,IFN参与疾病进程,IFN是细胞对各种不同的刺激(包括接触病毒)的反应所产生的一些特殊的蛋白质或糖蛋白,除了诱导产生抗病毒蛋白外,还可调节许多细胞功能[18]。在SLE病人中,免疫复合物诱导浆细胞样树突状细胞(pDCs)产生IFN,刺激B细胞分化为浆细胞,并随着中性粒细胞和髓系细胞的激活而发展为特异性组织(如肾脏)和全身炎症[19]。本研究发现的核心基因IFIT1、IFIT3、TRIM22、OAS1皆为IFN诱导基因,提示它们可能通过影响IFN参与LN发病。
核心基因IFIT1与IFIT3皆为含有四肽重复序列的干扰素诱导蛋白家族成员,属于IFN系统。IFIT所编码的蛋白受IFN诱导产生,并且主要由IFN-α/β诱导。IFIT家族成员通过多种机制调节免疫反应,限制病毒感染,包括限制病毒RNA翻译[20]。有研究显示,当IFIT1与IFIT3相互作用时,可以增强其抗病毒的活性[21]。研究表明,LN中IFIT1表达与足细胞损伤有关,尽管二者之间的机制尚不清楚,但IFIT1的表达与足细胞结构蛋白(包括F-肌动蛋白、肾素、podocin)之间的反向关联已被证明,提示IFIT1可能参与了LN的发病[22]。OAS1是由干扰素诱导并编码合成2’-5’-寡腺苷酸的核心基因,可激活潜在的核糖核酸酶L,从而导致病毒RNA降解并抑制病毒复制。除抗病毒外,已证明OAS1与SLE密切相关[23],并在一定程度上影响疾病活动度。本文结果与以上研究结果一致,进一步证明了这些基因在LN中具有重要作用。
位于MCODE子网络中核心的PLSCR1和核心基因TRIM22皆为干扰素诱导基因,可能介导干扰素的抗病毒作用,并与免疫应答密切相关。目前,尚无充足证据证明TRIM22在LN病程中具有直接作用,而PLSCR1是细胞激活过程中参与磷脂酰丝氨酸外化调控的关键分子,被证明是SLE血栓前倾向的重要因素之一[24]。本研究提示二者在LN中起正向调控作用,可能成为新的生物学靶点。
转录因子是调节基因表达的一组蛋白。本文研究结果显示,STAT1和STAT2是STAT家族的成员。该蛋白可以被各种配体激活,包括干扰素-α、干扰素-γ、表皮生长因子、血小板源生长因子和白细胞介素6。研究表明,STAT-Janus激酶信号通路对SLE至关重要,该通路激活导致STAT1的磷酸化和核移位,从而诱导细胞因子信号抑制物1和3的表达[25],导致细胞因子失调,这也是LN的重要标志。目前正在研究的小分子JAK抑制剂用于SLE治疗机制可能是由于它们抑制STAT蛋白磷酸化。本研究结果显示,STAT1和STAT2对LN致病基因具有调节作用,提示抑制由STAT介导的下游信号传导是LN有价值的治疗选择。
本文选择数据集为GSE32591,由于样本含量少,故在DEGs的富集分析中,未得到GO分析在细胞组分、分子功能层面上显著的功能富集, LN的发生发展在基因层面的机制需扩大样本量进一步研究。另一方面,该数据集的实验样本来源于美国人,与中国病人的基因表达有一定的差异,LN在不同人种间有不同的发病模式,因此,选择中国LN病人的数据进行基因层面的探索很有必要。
综上所述,LN在肾小球和肾小管间质中共有的DEGs有66个。GO分析和KEGG分析表明,DEGs在防御病毒、甲型流感中显著富集。在构建的PPI网络中预测得到4个核心基因和2个转录因子。本文结果为确定LN诊断和治疗的生物标记物提供了依据。
