N6-甲基腺嘌呤去甲基化酶抑制剂研究进展
2023-01-19高静,米雪,周琦,周君
高 静,米 雪,周 琦,周 君
(中国药科大学生命科学与技术学院,南京 211198)
RNA 修饰在生命活动和疾病的发生发展中发挥着重要作用。目前,已鉴定的RNA 修饰达150余种,这些修饰广泛分布于信使RNA(messenger RNA,mRNA)、核糖 体RNA(ribosomal RNA,rRNA)、转运RNA(transfer RNA,tRNA)、核内小RNA(small nuclear RNA,snRNA)和核仁小RNA(small nucleolar RNA,snoRNA)等各种RNA 上。RNA 修饰的种类繁多,不同类型的RNA 修饰的含量和功能存在较大差异[1]。其中RNA 甲基化修饰约占RNA 修饰的60%以上,是RNA 修饰的主要形式之一。甲基化修饰发生在RNA 上的位置主要是碱基的氮原子(N)、嘌呤和嘧啶的碳原子(C)、2'-OH 的氧原子(O)上。常见的RNA 甲基化修饰类型有N6-甲基腺嘌呤(m6A)、5-甲基胞嘧啶(m5C)、1-甲基腺嘌呤(m1A)、5-羟甲基胞嘧啶(hm5C)、假尿嘧啶(Ψ)等,它们在胞内行使不同的生物学功能。而m6A修饰作为其中具有代表性的修饰之一,也是目前研究最为透彻的一种RNA修饰类型[2]。
深入研究发现,m6A修饰及其效应蛋白与多种生理和病理过程密切相关[3]。例如,m6A 去甲基化酶FTO 的高表达与急性髓系白血病[4]、肺癌[5-6]等癌症发生发展密切相关,其另一去甲基化酶ALKBH5 也可通过影响m6A 修饰水平从而促进包括胶质母细胞瘤[7]和胃癌[8]在内的多种肿瘤进展,因此通过小分子靶向干预m6A 效应蛋白可作为抗肿瘤的新策略。
本文总结了有关m6A修饰的研究成果,阐述了其效应蛋白的类型、作用方式、生物学功能以及m6A 去甲基化酶FTO 和ALKBH5 抑制剂的开发进展,为全面了解m6A去甲基化酶作为疾病诊断和预后生物标志物提供了理论基础。
1 m6A甲基化修饰
1974年,Desrosiers 等[2]在 哺 乳 动 物 细 胞 的mRNA 中发现了m6A 的存在,即腺嘌呤第6 位氮原子上发生的单甲基化。但由于缺乏稳定的分析方法,m6A 的分布、功能以及作用机制研究很长时间处于停滞不前的状态。直到2011年,芝加哥大学何川教授团队报道了第1 个m6A 去甲基化酶——FTO[9],它可以去除mRNA 内部的m6A 修饰,揭示m6A是一种可逆化修饰,由此开创并引发表观转录组 学 的 研 究 热 潮。2012年,Dominissini 等[10]和Meyer 等[11]独立报道了全转录组水平m6A 的高通量测序方法(m6A-seq 或MeRIP-seq),为人们进一步研究m6A 奠定了非常坚实的基础。高通量测序研究结果表明,m6A 修饰是真核生物mRNA 中丰度最高的甲基化修饰形式,它广泛存在于病毒RNA、酵母、果蝇、植物及哺乳动物等真核生物中,并且m6A修饰位点附近的序列具有高度保守性,倾向于发生在RRACH(R=G/A,H=A/C/U)序列的腺嘌呤上,这些位点主要分布在RNA 的3'UTR、终止密码子或者长外显子附近,且该分布特征在人类及小鼠中是高度保守的[12]。
研究表明,m6A修饰是一种动态可逆的调节方式,它受m6A 甲基转移酶(m6A writers)和去甲基化酶(m6A erasers)的共同调控,能够被m6A 识别蛋白(m6A readers)选择性的识别结合,从而在转录后调控基因的表达,进而发挥相应的生物学功能[3]。目前已发现的甲基转移酶复合物的主要成分包括
METTL3、METTL14[13]、WTAP[14]、KIAA1429(VIRMA)[15]、RBM15[16]、HAKAI[17]、ZC3H13(KIAA0853)[18]和METTL16[19]等,它们能够催化S-腺苷甲硫氨酸(S-adenosyl methionine,SAM)的甲基转移到腺嘌呤的第6 位氮原子上。而FTO[9]和ALKBH5[20]作为去甲基化酶能动态可逆地去除靶RNA的m6A 甲基化修饰。m6A 修饰的生物学功能主要是通过m6A识别蛋白发挥调控作用的,目前已知的识别蛋白主要有YTH 结构域蛋白(YTHDF1[21]、YTHDF2[22]、YTHDF3[23]、YTHDC1 和YTHDC2)、核不均一核糖核蛋白HNRNP 家族蛋白(HNRNPA2B1[24]、HNRNPC 和HNRNPG)和胰岛素样生长因 子2 mRNA 结 合 蛋 白IGF2BP 家 族 蛋 白[25](IGF2BP1、IGF2BP2、IGF2BP3)。通过m6A 识别蛋白的结合,不仅可以影响mRNA 的稳定性、促进蛋白翻译效率、调控mRNA 与miRNA 的剪接和促进mRNA 出 核[26]。m6A 也 可以干 扰底物RNA 的结构,作为“结构开关”调控HNRNPC 与RNA 的结合[27]。此外,m6A 参与了多种生物学过程[28],如干细胞分化、细胞分裂、配子发生和生物节律等,以及多种疾病的发生[29],包括肿瘤、肥胖、神经性疾病和不育等,在个体发育及生理病理调控中发挥着重要的作用。
2 m6A去甲基化酶
AlkB家族作为铁(Ⅱ)和酮戊二酸(2-OG)依赖性加氧酶是一种重要的核酸脱甲基酶,图1展示了9 个AlkB 人类同源蛋白的基因结构,通过保守性分析发现它们都拥有“HXD…H”(X 表示任何氨基酸)和“R…R”两个基序,已知这两个基序分别在铁(Ⅱ)结合以及酮戊二酸结合过程中起到重要作用。其中FTO 和ALKBH5 是值得关注的两个m6A去甲基化酶。

图1 人类AlkB家族的基因结构图以及保守性分析
2.1 FTO去甲基化酶
早在2007年就有文献报道过FTO 的氧化去甲基化作用[30],后续又证明甲基化的RNA 是FTO 的首选底物[31]。而2011年发表在Nature Chemical Biology杂志上的研究工作揭示了FTO 在体外针对RNA 中丰富的m6A 残基具有高效的氧化去甲基化活性,证实核RNA 中的m6A 是FTO 的主要生理底物[9]。
如图2 所示,FTO 在不用物种中的代表性转录本含有代表铁(Ⅱ)结合的“HXD…H”基序和代表酮戊二酸结合的“R…R”基序。

图2 不同物种中脂肪含量与肥胖相关蛋白(FTO)的的基因结构图以及保守性分析
有实验发现,在脂肪形成过程中FTO 表达和m6A 水平呈负相关,FTO 消耗会阻碍分化,只有催化活性的FTO 才能恢复脂肪生成[32]。FTO 能够一定程度上促进急性髓系白血病[4]、非小细胞肺癌[5]、人乳腺癌[33]和膀胱癌[34]等的发展。另外,FTO 抑制与抗PD-1阻断相结合可能会降低黑色素瘤免疫治疗的耐药[35]。与相邻的正常组织相比,宫颈鳞状细胞癌组织中FTO 能调节β-catenin 的表达,在体外和体内增强化疗-放疗的抗性[36]。
2.2 ALKBH5去甲基化酶
ALKBH5 是在2013年被发现的第2 个m6A 去甲基化酶[37]。该研究证实其是另一种哺乳动物去甲基化酶,可在体外和体内去除核RNA(主要是mRNA)上的m6A修饰[37]。
类似于FTO,ALKBH5转录本在不同物种中的代表性转录本(图3)也含有代表铁(Ⅱ)结合的“HXD…H”基序和代表酮戊二酸结合的“R…R”基序。
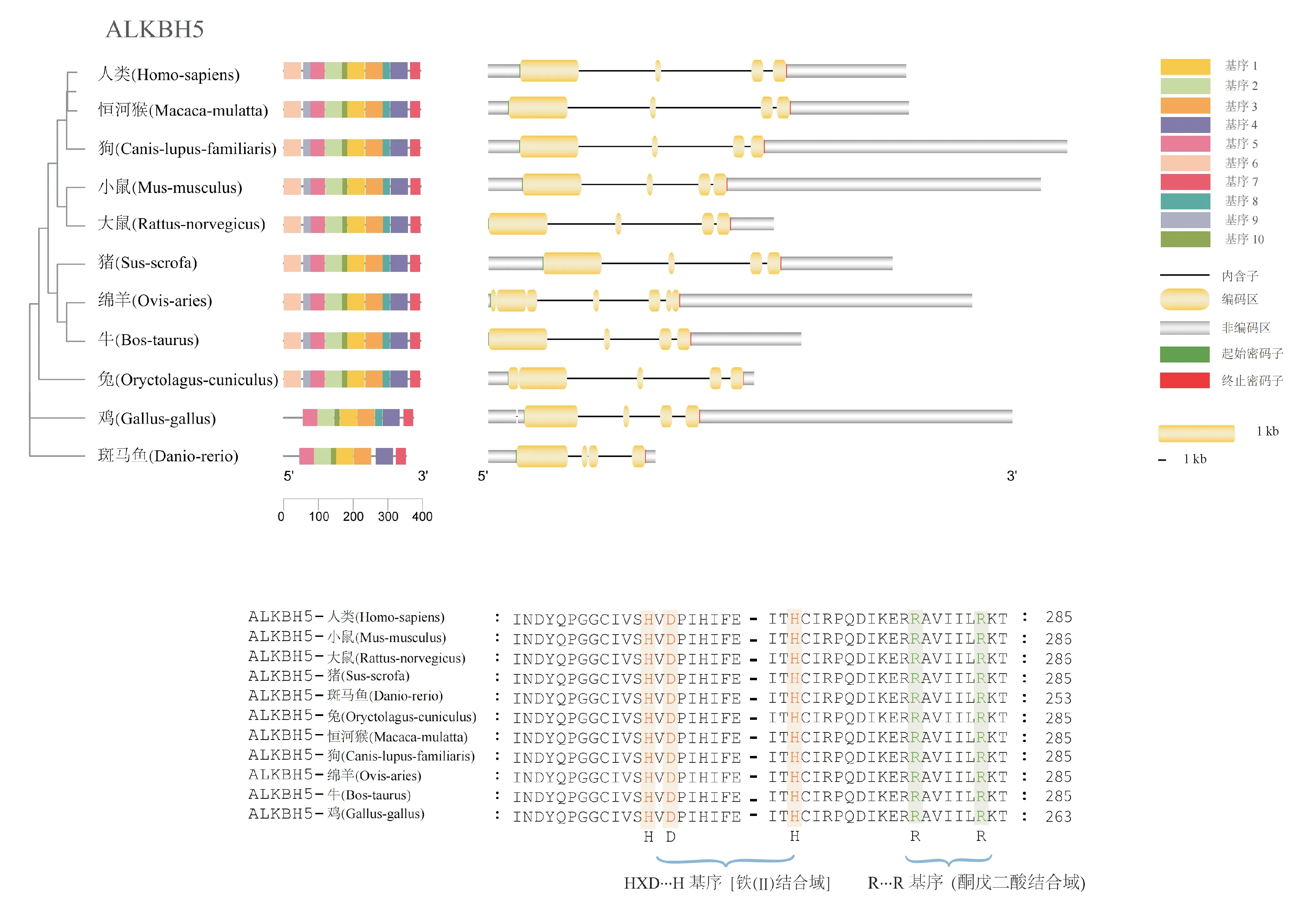
图3 不同物种中AlkB同源蛋白5(ALKBH5)的基因结构图和保守性分析
ALKBH5 在组织中广谱表达。其在睾丸中特别丰富,能够影响小鼠精子发生和生育能力[38]。抑制ALKBH5 能减少自噬通量并促进缺氧/复氧(hypoxia/reoxygenation,H/R)处理的心肌细胞的凋亡[39]。此外,ALKBH5 能够抑制胃癌细胞[40]、肺腺癌细胞[41]和上皮性卵巢癌细胞[42]等多种细胞的生长。另有报道称ALKBH5 的缺失使黑色素瘤对其的免疫治疗增敏[43],有助于免疫治疗的疗效,可作为一个潜在的治疗靶点。
3 m6A去甲基化酶抑制剂的开发
3.1 FTO去甲基化酶抑制剂
FTO 是全基因组关联研究中发现的第一个肥胖风险基因,此外,它还是第1 个被发现的可逆的m6A 去甲基化酶。作为非血红素双加氧酶超家族中的成员之一,FTO 能够催化铁(Ⅱ)和α-酮戊二酸(2-OG)依赖的底物氧化去甲基化过程,因此,从作用机制上来说,FTO抑制剂可分为金属离子螯合剂、2-OG类似物和底物竞争性抑制剂3类。
通用型抑制剂N-草酰甘氨酸(N-oxalylglycine,NOG)与2-OG 有类似结构,可以竞争性抑制FTO与下游底物的结合[44]。研究表明,环状和非环状2-OG 类似物都具有抑制FTO 的作用。然而由于它们较低的特异性和选择性,难以在某些因FTO高表达导致疾病进展的肿瘤中作为靶向治疗药物。因此研发出高选择性和特异性的FTO 抑制剂成为此后的研究重点。目前,已有研究报道了一些FTO 抑制剂(表1),其化学结构式如图4所示。

图4 m6A去甲基化酶FTO抑制剂的化学结构式

表1 已开发的N6-甲基腺嘌呤(m6A)去甲基化酶FTO抑制剂
3.1.1 大黄酸(rhein) 2012年,上海药物研究所通过高通量虚拟筛选和生化分析等方法,将天然产物大黄酸(rhein,化合物1)确定为第1 个特异性较高的FTO 抑制剂[45]。根据生物物理实验分析以及动力学研究发现,大黄酸既不是2-OG 结构类似物,也不是结合2-OG 辅助因子的金属离子螯合剂,而是通过与FTO 底物竞争,特异地结合其催化结构域,从而抑制FTO去甲基化酶功能。
首先,限制性核酸内切酶消化、等温滴定量热法、荧光偏振法和液相色谱分析等体外实验验证了大黄酸的抑制活性。其次,在细胞实验中,大黄酸也被证明能够提高人神经瘤细胞系BE(2)-C 中总mRNA的m6A修饰水平,并且大黄酸对BE(2)-C细胞并未表现出明显的细胞毒性,这为日后大黄酸在细胞实验研究和临床应用中增加了更多的可能性。然而大黄酸对AlkB 亚家族的选择性很小[46]。
3.1.2 甲氯芬那酸(MA)和((2E)-4-[N‘-(4-苄基吡啶-3-羰基)-肼基]-4-氧代丁-2-烯酸) 为了筛选FTO 的选择性抑制剂,上海药物所研究团队于2015年通过高通量荧光偏振(FP)方法比较了各种药物化合物存在下m6A-ssDNA 结合FTO 和AlkBH5 的差异[47]。结果发现非甾体抗炎药甲氯芬那酸(MA,化合物2)以剂量依赖的方式抑制含有m6A 的ssDNA 或ssRNA 的FTO 去甲基化,却不能与ALKBH5 蛋白结合,也不竞争ALKBH5 与ssDNA 的结合。进一步的抑制机制研究显示,MA既不是2-OG 的化学模拟物,也不是铁的螯合剂,它与FTO 结合形成一个发夹基序。该基序作为核酸识别盖(nucleotide recognition lid,NRL)的一部分,可用于提供MA 和FTO 之间额外的相互作用,而ALKBH5 恰好缺乏NRL 部分的发夹基序。因此MA 是体外FTO 去甲基化的高选择性抑制剂,并且针对MA/FTO 结构化合物可进行强效FTO 抑制剂的结构引导设计。
同年,新加坡国立大学的TOH 研究团队[48]基于AlkB酶的核苷酸结合位点的固有结构差异的原理,设计合成小分子化合物来更有效地选择性抑制AlkB 酶。在筛选的数十种化合物中,((2E)-4-[N'-(4-苄基吡啶-3-羰基)-肼基]-4-氧代丁-2-烯酸)(化合物3)被确定为一种有效的FTO 选择性抑制剂。它对FTO 的选择性是其他AlkB 亚家族的30~130 倍。晶体学分析显示,((2E)-4-[N'-(4-苄基吡啶-3-羰基)-肼基]-4-氧代丁-2-烯酸)同时占据FTO蛋白中核苷酸与2-OG的结合位点。细胞实验进一步表明,它的乙酯衍生物能够抑制细胞中m6A去甲基化酶的活性。这是首次报道的一种对特定的AlkB 亚家族成员具有选择性的抑制剂,这种选择性为其作为功能探针和治疗先导物提供了新思路。
3.1.3 N-CDPCB 和CHTB N-CDPCB(1a,化合物4)是由郑州大学的常俊标研究团队于2015年设计得到的新型FTO 小分子抑制剂[49]。通过鉴定人FTO 与N-CDPCB 的晶体结构,发现其与报道的FTO 特异性抑制剂MA 的结构有部分重叠,并且发现N-CDPCB与FTO蛋白的相互作用主要是通过范德华力、疏水作用力和氢键来实现的,揭示了FTO抑制剂的一个新的结合位点以及它们相互作用的结构基础。
2016年,利用相同的设计方法,该研究团队又发现了另一种FTO 抑制剂CHTB(化合物5)[50],它的作用方式与N-CDPCB相似,也是通过范德华力、疏水作用力和氢键来实现对FTO 蛋白的特异性识别的。结构分析显示CHTB 占据了FTO 蛋白活性位点的L 形空腔。新的结合位点的识别为进一步开发选择性和强效的FTO 抑制剂提供了新的机会,这有望为治疗肥胖或肥胖相关疾病的新治疗靶点提供信息。
3.1.4 天然化合物根赤壳菌素(radicicol) 2018年,郑州大学的常俊标研究团队在进一步研究该新结合位点的构效关系后发现,4-氯-1,3-二醇基团是N-CDPCB(1a)、CHTB 以及它们的类似物的共同结构基序。因此,研究人员根据N-CDPCB/FTO复合物中发现的新的结合位点,对具有4-氯-1,3-二醇基团的化合物进行了基于结构的虚拟筛选,同时利用相似性搜索和活性分析,进而确定了天然化合物根赤壳菌素(radicicol,化合物6)是一种有效的FTO 抑制剂[51]。实验结果显示,它对FTO去甲基化活性呈剂量依赖性抑制,半抑制浓度为16.04 µmol/L。并且两者之间的相互作用主要是熵驱动的。晶体结构分析表明,根赤壳菌素在FTO 结合位点采用L 形构象,并与之前发现的FTO小分子抑制剂N-CDPCB占据相同的位置。该抑制剂的发现为设计更有效的化合物来抑制FTO 的活性提供了新的信息。
3.1.5 FB23 和FB23-2 继2015年筛选出MA 抑制剂之后,上海药物所Huang 等[52]在2019年进一步优化合成了MA 衍生物FB23(化合物7)和FB23-2(化合物8)。基于结构导向的合理设计开发的这两种FTO 抑制剂,即FB23 和FB23-2,通过占据底物结合位点直接与FTO 结合,选择性地抑制FTO的m6A去甲基化酶活性。
体外实验结果显示,FB23-2 能显著抑制人急性髓系白血病(AML)细胞系细胞和原代母细胞AML 细胞的增殖,促进其分化或凋亡。此外,FB23-2也能显著抑制异种移植小鼠中的人AML细胞系和原代细胞的进展。这可能对通过靶向表观转录组RNA 甲基化进行癌症治疗,产生广泛的影响。
3.1.6 恩他卡朋(entacapone) 2019年,北京生命科学研究所的研究团队使用了一种基于结构的对美国食品药品监督管理局FDA 批准的药物进行虚拟筛选的方法,进而将儿茶酚-O-甲基转移酶(catechol-O-methyltransferase,COMT)抑制剂恩他卡朋(化合物9)确定为一种潜在的FTO 抑制剂[53]。它最初被FDA 批准是作为左旋多巴和卡比多巴联合治疗帕金森病的辅助治疗。通过结构和生化研究,发现恩他卡朋在体外直接结合FTO 并抑制FTO 活性。它可以降低饮食诱导的肥胖小鼠的体重和空腹血糖浓度。此外,RNA-seq结果还鉴定出转录因子叉头框蛋白O1(forkhead box protein O1,FOXO1)mRNA 是作为FTO 的直接底物,并证明了恩他卡朋在FTO-FOXO1调节轴对小鼠肝脏糖异生和小鼠脂肪组织生热的调节作用中具有重要影响。
3.1.7 CS1(NSC337766)和CS2(NSC368390) 虽然m6A 修饰与各种类型癌症的起始、进展、维持和耐药性密切相关,但目前针对m6A 调节剂进行癌症治疗的有效抑制剂的开发仍处于起步阶段。2020年,陈建军教授团队首先对来自美国国家癌症研究所的上万种化合物进行了基于结构的虚拟筛选以及后续验证分析,鉴定出两种有效的小分子化合物CS1(化合物10)和CS2(化合物11)[54],它们可通过靶向FTO 进而抑制癌症干细胞的维持和免疫逃避。
CS1 和CS2 在抑制急性髓系白血病细胞活力和FTO 去甲基酶活性方面表现出强大效果,通过靶向FTO 并占据催化活性位点进而干扰FTO 与m6A 修饰RNA 的结合,有效地抑制其m6A 去甲基化酶活性。实验结果显示,CS1 和CS2 通过抑制FTO 活性和干扰FTO 的信号通路,比如激活凋亡信号通路和抑制MYC 通路,进而发挥抗白血病作用。与之前报道的FB23-2 等FTO 抑制剂相比,CS1 和CS2 在抑制急性髓系白血病细胞活力方面表现出10 ~ 30 倍的功效,表明它们的疗效大大提高。
3.1.8 谷胱甘肽生物印迹纳米复合材料(GNPIPP12MA) 尽管近年来一些小分子FTO 抑制剂的开发取得了很有前景的进展,但由于生物功能温和、不良反应和对白血病干细胞的敏感性和特异性较低,其临床潜力仍然有限。2022年Cao等[55]研究开发了负载FTO 抑制剂的谷胱甘肽GSH生物印迹纳米复合材料(GNPIPP12MA),它通过靶向FTO/m6A 通路协同GSH 消耗来增强抗白血病发生。
实验结果显示,GNPIPP12MA 可选择性靶向白血病母细胞,尤其是白血病干细胞,通过破坏细胞内氧化还原状态诱导铁死亡。此外,GNPIPP12MA 增加了白血病干细胞中的m6A 修饰,降低了转录水平。GNPIPP12MA 通过增加细胞毒性T 细胞的浸润来增强抗白血病免疫,从而增强PD-L1 阻断的疗效。这项研究为靶向m6A 甲基化的GSH 生物印迹纳米平台提供了新的思路,该平台可作为一种协同治疗肿瘤干细胞的策略,有望应用于临床。
3.1.9 小分子抑制剂18097 2022年,研究人员通过虚拟筛选、结构优化和生物测定等方法开发出了一种新的FTO 小分子抑制剂18097(化合物12)[56]。
小分子抑制剂18097 通过与FTO 活性位点结合,进而抑制细胞周期过程和癌细胞迁移。可以对乳腺癌细胞的表观转录组进行重编程,尤其是P53通路相关基因的重编程。小分子抑制剂18097通过招募IGF2BP1 来增加mRNA 的稳定性,进而激活P53信号通路。此外,它还抑制了细胞脂肪生成,通过下调PPARγ 和C/EBPα/β mRNA 的稳定性来降低二者的表达。动物实验证实,小分子抑制剂18097能显著抑制乳腺癌细胞的生长和肺定植。综上所述,FTO 可以作为一种潜在的药物靶点,小分子抑制剂18097是一种特异性的、有效的FTO 抑制剂,可在体内外抑制实体肿瘤细胞,尤其对乳腺癌细胞的体内转移有抑制作用,因此可以作为一种潜在的抗乳腺癌药物。
3.2 ALKBH5去甲基化酶抑制剂
与FTO 不同的是,ALKBH5 直至2013年才初次被报道具有m6A 去甲基化酶作用[37],并且它也是Fe(Ⅱ)/2-OG依赖性双加氧酶。而有关ALKBH5抑制剂的研究一直鲜有报道(表2),目前已开发的ALKBH5抑制剂的化学结构式见图5。

表2 已开发的m6A去甲基化酶ALKBH5抑制剂

图5 m6A去甲基化酶ALKBH5抑制剂的化学结构式
3.2.1 柠檬酸盐 2014年,Xu 等[57]发现TCA 循环中间体柠檬酸盐可以作为ALKBH5 温和型抑制剂,IC50约为488 µmol/L,具有较弱的抑制效应。柠檬酸与ALKBH5 的结合方式与在FTO 中观察到的不同。在FTO-柠檬酸复合物中,柠檬酸取代了2-OG,而金属离子仍然完好无损。但在ALKBH5-柠檬酸复合物中,柠檬酸同时占据了金属离子和2-OG。虽然还需要对ALKBH5复合物的结构进行进一步的研究,但通过初步分析ALKBH5 结合底物的特异性环状结构,不仅可以为ALKBH5对ssRNA去甲基化的分子机制提供见解,确定柠檬酸盐作为ALKBH5 的温和型抑制剂,同时也为ALKBH5的高特异性抑制剂开发奠定了基础。
由于该化合物对U87-MG 细胞的影响似乎并不依赖于其钠通道阻断能力,因此通过使用SPILLO-PBSS 软件确定了MV1035 的替代脱靶相互作用。在MV1035排名最高的脱靶点中,重点关注的是RNA 去甲基化酶ALKBH5 酶,该酶在癌症中发挥了关键的肿瘤抑制作用。体外实验结果也表明,MV1035可以靶向m6A去甲基化酶ALKBH5,从而提高m6A 水平。此外结果还进一步表明,CD73 可能是作为下游靶蛋白参与MV1035 抑制ALKBH5,降低U87细胞系迁移和侵袭性的途径。3.2.3 化合物20m 2022年,Fang 等[59]报道了一类新的含有1-芳基-1H-吡唑支架的ALKBH5 抑制剂。通过基于荧光极化的筛选、结构优化和构效关系分析发现,化合物20m(化合物14)的荧光极化作用最强,IC50为0.021 µmol/L,且化合物20m对ALKBH5 比对FTO 以及其他AlkB 亚家族成员具有更高的选择性。细胞热转移实验(CETSA)结果显示,化合物20m 可有效稳定HepG2 细胞中的ALKBH5。斑点印迹实验表明,化合物20m可提高完整细胞的m6A水平。综上所述,化合物20m是一种高效选择性的具有细胞活性的ALKBH5 抑制剂,可作为一种多功能的化学探针来探索ALKBH5 的生物学功能。
4 m6A去甲基化酶抑制剂的应用与展望
越来越多的研究表明,m6A去甲基化酶在多种疾病的发生发展中发挥着重要作用,其中去甲基化酶FTO 与急性髓系白血病[4]、胰腺癌[60]和肺癌[6]等癌症发生发展密切相关,其高表达会影响肿瘤细胞的生长和增殖、肿瘤干细胞的自我更新[61]、肿瘤转移以及导致不良的肿瘤预后[35]。去甲基化酶ALKBH5 可通过影响m6A 修饰水平从而促进包括胶质母细胞瘤[7]和胃癌[8]在内的多种肿瘤进展。因此,已经有越来越多的去甲基化酶抑制剂被开发并应用于抗肿瘤治疗。FTO 是目前研究和发展最多的m6A 相关酶,而最新报道的FTO 抑制剂也达到了很高的活性和选择性,并展现出一定的临床应用潜能。随着各项技术的不断突破,ALKBH5抑制剂的筛选也处于稳步发展的过程中。
然而,大部分化合物仍处于生化或细胞实验验证阶段,部分化合物即使经过了小鼠体内验证,在人体环境中,其抑制效率和安全性维持仍未可知,并且对于抑制剂调控肿瘤的具体作用机制也未能清晰阐述,远不能满足新药的临床前研究推进。因此,高效高选择性抑制剂的开发和研究仍有待进一步深入。
对于已开发的小分子化合物,还需要进一步系统研究其调控肿瘤的机制,并通过进一步优化,以提高其生物利用度、抑制效果和治疗效果。同时分析此类抑制剂应用于各种肿瘤之间的异同,对于肿瘤的诊疗具有重要的理论意义和应用价值。此外,通过药物化学等手段筛选新的更高活性和选择性的小分子抑制剂,开发高特异性抑制剂有助于肿瘤研究中甲基化修饰功能的探究。这些筛选出的药物有望成为有用的工具分子,帮助研究人员进一步探索m6A 相关生物过程的机制,同时对于肿瘤临床治疗也有着积极意义。
