注射用美罗培南聚合物杂质分析方法的比较研究
2023-01-16程继业周震宇金伟斌顾晓风
程继业,姚 玲,周震宇,金伟斌,顾晓风
苏州市药品检验检测研究中心,苏州 215104
美罗培南(meropenem)是一种半合成的广谱碳青霉烯类抗生素,具有抗菌活性强、对β-内酰胺酶稳定、副作用小等特点,对革兰阳性菌和革兰阴性菌均有较强的抗菌活性[1],临床用于混合感染和各种严重感染[2-5],尤其多用于入住重症监护病房(Intensive Care Unit,ICU)需进行高强度的抗感染治疗的患者[6]。本品对中枢神经系统及肾脏的毒副作用较小,临床应用中不良反应发生率低,但本品属于β-内酰胺类抗生素,在水溶液中不稳定,易产生聚合物杂质,国内外研究表明,包括聚合物在内的高分子杂质与β-内酰胺类抗生素的变态反应有密切联系[7-9],因此建立聚合物杂质的检测方法对保障本品质量十分重要。
《中国药典》2020年版、《美国药典》2021年版均收载了该品种,但均未单独设置聚合物检查项[10-11]。《欧洲药典》10.0版在美罗培南原料的有关物质项下,将美罗培南二聚体(杂质B)作为特定杂质控制[12]。有文献报道用Sephadex G-10葡聚糖凝胶色谱柱、TSK G2000SW/600以及G2000SWxl凝胶柱对美罗培南原料和制剂的聚合物杂质进行测定[13-15]。G-10色谱法操作耗时,定量准确度较低,已逐渐被高效凝胶色谱法代替[14,16]。相比G-10柱,2个G2000高效凝胶柱方法实现了对美罗培南聚合物的分离,并通过质谱法对分离得到的主要杂质结构进行推断,为美罗培南二聚体和异构体杂质,由于缺少杂质对照品,选择以主成分外标法[14]或自身对照法[15]对聚合物总量进行控制。
本品为2021年国家评价性抽验品种,产品来自包括原研厂家在内的11个生产厂家,涉及包括《中国药典》在内的7个质量标准,只有YBH00182011、YBH02582015、YBH02162016 3个标准收载了聚合物检查方法,在对应企业产品分析中,发现3个标准存在分析时间长、方法专属性差、色谱柱效低、分离度差等问题[17-19]。本文在上述标准基础上选择更高效的TSK G2000 SWxl色谱柱,优化色谱条件,建立美罗培南聚合物杂质的分析方法,对国内上市的11家企业的33批产品进行分析,并与基于《中国药典》2020版及《欧洲药典》10.0版开发的有关物质方法测定结果进行比较,探讨高效凝胶色谱法(HPSEC)和反相高效液相色谱法(RP-HPLC)在测定抗生素聚合物方面的优劣,为本品国家药品标准的提高提供依据。
1 仪器与试药
1.1 仪器
Waters e2695高效液相色谱仪(配DAD检测器,Empower Pro工作站);梅特勒托利多XSE205DU电子天平;Millipore Milli-Q超纯水仪。
1.2 试药
美罗培南对照品(中国食品药品检验研究院,批号130506,按美罗培南计质量分数为86.8%);美罗培南杂质A对照品(批号20200723,质量分数为97.95%),美罗培南杂质B对照品(批号20201110,质量分数为95.49%),均购自杭州卓越科技生物有限公司;注射用美罗培南(共33批,11个厂家,A~K);三乙胺(Fisher Chemical,色谱纯);乙腈(Honeywell,色谱纯);二水合磷酸二氢钠、十二水合磷酸氢二钠、磷酸(国药集团化学试剂有限公司,均为分析纯);水为超纯水。
2 方法与结果
2.1 HPSEC色谱条件与测定法
色谱柱为TOSOH TSK G2000 SWxl (30 cm×7.8 mm, 5 μm);流动相为0.01 mol·L-1磷酸氢二钠溶液-0.01 mol·L-1磷酸二氢钠溶液(61∶39,pH7.0);流速为0.8 mL·min-1;柱温为30 ℃;检测波长为254 nm;进样量为20 μL。
取本品内容物,相当于美罗培南100 mg,置于50 mL量瓶中,加水适量使溶解并稀释至刻度,摇匀,用0.45 μm微孔滤膜滤过,作为供试品溶液,溶液应临用新制。
测定法:取供试品溶液室温放置1 h,精密量取20 μL,注入液相色谱仪中,记录色谱图,美罗培南杂质峰(相对保留时间约为0.8)与美罗培南峰的分离度应不小于3.0,美罗培南峰的理论板数应不低于5 000,色谱图见图1。精密量取供试品溶液,注入液相色谱仪,杂质峰按峰面积归一化法计算。
2.2 RP-HPLC色谱条件与测定法
色谱柱为SUMIPAX ODS A-212(150 cm×6 mm,5 μm);流动相为1 mL·L-1三乙胺溶液[取三乙胺1.0 mL,加水900 mL,用磷酸溶液(1→10)调pH值至5.0±0.1,加水稀释至1 000 mL]-乙腈(93.5∶6.5);流速为1.6 mL·min-1;柱温为40 ℃;检测波长为220 nm;进样量为10 μL。
取本品内容物,用1 mL·L-1三乙胺溶液溶解并定量稀释制成5 mg·mL-1的溶液,作为供试品溶液,溶液应临用新制。精密量取供试品溶液1 mL,置于200 mL量瓶中,用1 mL·L-1的三乙胺溶液稀释制至刻度,摇匀,作为对照溶液。
测定法:取美罗培南对照品适量,加1 mL·L-1的三乙胺溶液溶解并定量稀释成0.5 mg·mL-1的溶液,置于水浴中加热1 h,放冷,精密量取10 μL,注入液相色谱仪,美罗培南峰保留时间约为7 min,出峰顺序依次为美罗培南开环物(杂质A)、美罗培南与美罗培南二聚体(杂质B),杂质A与美罗培南峰的分离度应符合要求。再精密量取供试品溶液注入液相色谱仪,记录至主成分保留时间的3.5倍。如有杂质峰,按主成分自身对照法计算,其中杂质A 结果乘以1.6。
2.3 HPSEC法专属性实验用溶液配制
空白辅料溶液:本品处方中美罗培南与碳酸钠的比例约为500∶104,据此配制空白辅料溶液。取碳酸钠适量,加水溶解制成0.04 mg·mL-1的溶液,用0.45 μm微孔滤膜滤过。
酸破坏溶液:称取美罗培南对照品约200 mg,置于100 mL量瓶中,用水溶解并稀释至刻度,摇匀,作为储备液。量取储备液10 mL,加入1 mL 0.1 mol·L-1盐酸溶液,室温放置100 min,加入0.1 mol·L-1氢氧化钠溶液至pH 7,用0.45 μm微孔滤膜滤过。
碱破坏溶液:量取储备液10 mL,加入1 mL 0.1 mol·L-1氢氧化钠,室温放置15 min,加入0.1 mol·L-1盐酸溶液溶液至pH 7,同法滤过。
氧化破坏溶液:量取储备液10 mL,加入1 mL体积分数为30%过氧化氢溶液,室温放置30 min,同法滤过。
高温破坏溶液:量取储备液10 mL,水浴60 ℃,保温放置4 h,放冷,同法滤过。
光照破坏溶液:量取储备液10 mL,置于光照实验箱中,可见光4 500±500 lx/紫外光200 W·hr·m-2条件下放置20 h,同法滤过。
2.4 HPSEC法的专属性考察
本次国评品种中,3个企业适用的国家标准YBH00182011、YBH02162016、YBH02582015指定了不同的凝胶色谱柱:1)TSKG2000 SW/600(600 mm×7.5 mm,10 μm)、2)Shodex SUGAR ks-801(300 mm×8.0 mm,6 μm)、3)赛分Cef-SEC(300 mm×7.8 mm,5 μm),对应的典型色谱图见图2,3个标准均存在分析时间长、检测结果偏高、方法专属性差和色谱柱效低的问题。综合实验多种色谱柱后,本研究选择填料粒径更小、柱效更高的TSK G2000 SWxl (300 mm×7.8 mm,5 μm) 进行方法优化并验证。分别取2.3项下空白辅料溶液及破坏溶液按HPSEC法色谱条件分析,记录色谱图。
①空白辅料在美罗培南及相关杂质处无干扰峰;②各种破坏条件均容易使美罗培南产生杂质,杂质分布于保留时间(RT)10 min至美罗培南主峰附近。③酸、碱、高温、光照条件下均产生RT 11.9 min的主要杂质峰。用DAD检测器获得紫外光谱,发现酸、碱、高温条件下的峰具有相同的光谱,而光照条件下不同,说明在RT 11.9 min处可能存在不同的杂质。④氧化破坏试验产生杂质较复杂,超出了色谱柱的分离能力,导致杂质与美罗培南不能完全分离,其余破坏条件用DAD检测器对美罗培南主峰质量分数检查,峰质量分数符合要求。见图3。

注:美罗培南与杂质峰的分离度为4.1,美罗培南峰的理论板数为13 901。

注:A.(国家药品标准YBH00182011,色谱柱TSK G2000 SW/600); B.(国家药品标准YBH02582015,色谱柱Shodex SUGAR ks-801); C.(国家药品标准YBH02162016,色谱柱赛分Cef-SEC)。
2.5 美罗培南特定杂质色谱峰位确认
由于抗生素聚合物杂质制备难度较大,目前尚无法定对照品。通过对本品杂质研究文献及国际药典的查阅,确定美罗培南杂质A及杂质B是主要的相关杂质,基本信息见表1。商业途径获得了这2个杂质对照品,并对其在色谱过程中的峰位进行确认。
分别称取杂质A、B适量,用水溶解制成0.04 mg·mL-1的溶液,按2.1项下方法进样分析。结果杂质A与杂质B保留时间基本重合(11.9 、11.7 min),无法分离,也解释了在样品测试时不同厂家产品该位置处的色谱峰形不一致,可能由杂质相对含量不同所致。杂质A、杂质B紫外光谱见表1,推测酸、碱、高温条件下11.9 min的杂质可能为杂质A,光照条件下11.9 min的杂质与杂质B的紫外光谱存在差异,且质量分数检查显示大于阈值,表明该峰位处存在多种成分。
发现本方法的专属性问题后,我们又对3个法定方法的分离能力进行了确认,结果情况相同,即法定方法均不能分离美罗培南的2个主要降解杂质,方法拟控制的聚合物杂质实际是二聚体和小分子杂质总量。查阅至2022年1月美罗培南聚合物研究的文献,普遍认为分子排阻色谱分析美罗培南主峰前的杂质峰是聚合物,均未述及小分子杂质存在以及聚合物峰质量分数问题,可以认为,目前使用HPSEC进行美罗培南聚合物分析的方法存在一定专属性缺陷。

注:A.酸破坏;B.碱破坏;C.氧化破坏;D.高温破坏;E.光照破坏。
如前所述,本品种7个国家标准中,YBH00182011、YBH02582015、YBH02162016 收载了聚合物项目,且使用的色谱柱类型、定量方法各不相同,为了以相同方法和尺度对各厂家样品进行评价,我们仍使用新建的方法,以杂质总量为指标,对方法其他参数进行验证并评价各批次产品。
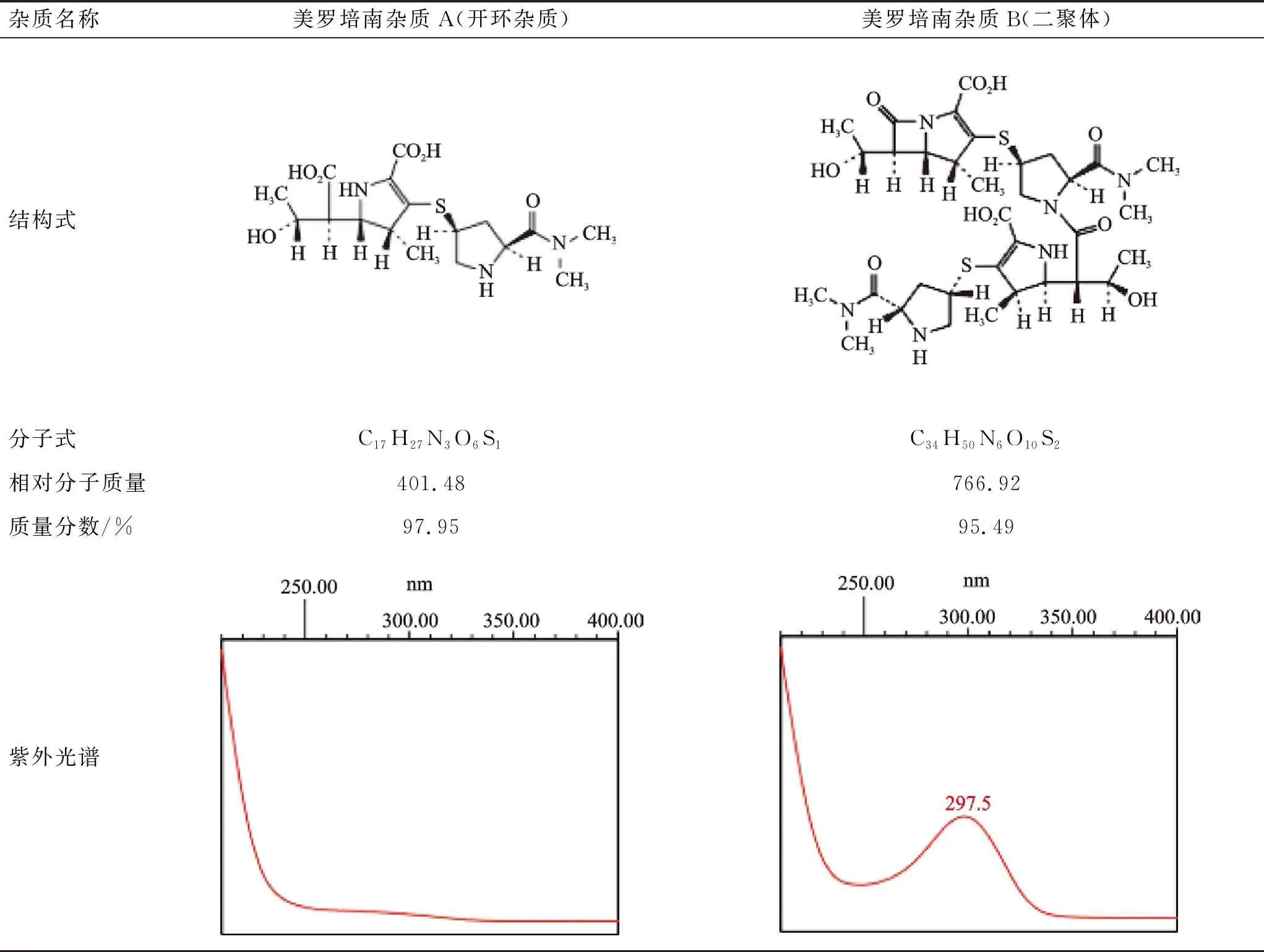
表1 美罗培南杂质A、杂质B的基本信息
2.6 HPSEC法相关方法学验证
2.6.1溶液稳定性 取供试品溶液,室温放置,不同时间分析,考察总杂质4 h的变化情况,结果总杂质含量由0.14%升高到0.40%,说明水溶液不稳定,应临用新制。
2.6.2方法重复性 取1批产品,按2.1项下方法制备5份供试品,进样分析。结果杂质峰保留时间相对标准偏差(RSD)为0.1%,与主峰分离度RSD为1.1%,杂质含量标准偏差(SD)为0.03,方法重复性好。
2.6.3线性与杂质A、杂质B校正因子的测定 取美罗培南对照品,按2.1项下方法,配制系列溶液(相当于供试品溶液中美罗培南质量浓度的0.1%~150%);取美罗培南杂质A、杂质B对照品,同法配制系列溶液(相当于供试品溶液中美罗培南质量浓度的0.1%~2%),分别进样分析,记录色谱图,以峰面积对其质量浓度进行线性回归,得线性方程,并计算校正因子。
美罗培南:y=12 103.0x-47 992.7,线性范围为1.82~2 724.65 μg·mL-1,r=1.000 0。
杂质A:y=1 805.1x-597.7,线性范围为2.05~41.00 μg·mL-1,r=1.000 0;校正因子为6.705。
杂质B:y=5 609.2x-3 536.7,线性范围为2.01~40.20 μg·mL-1,r=1.000 0;校正因子为2.158。
2.6.4检测限与定量限 分别取美罗培南、杂质A、杂质B对照品溶液,逐级稀释,依法测定,按信噪比约3和10时的待测物质量浓度作为检测限和定量限。结果见表2。
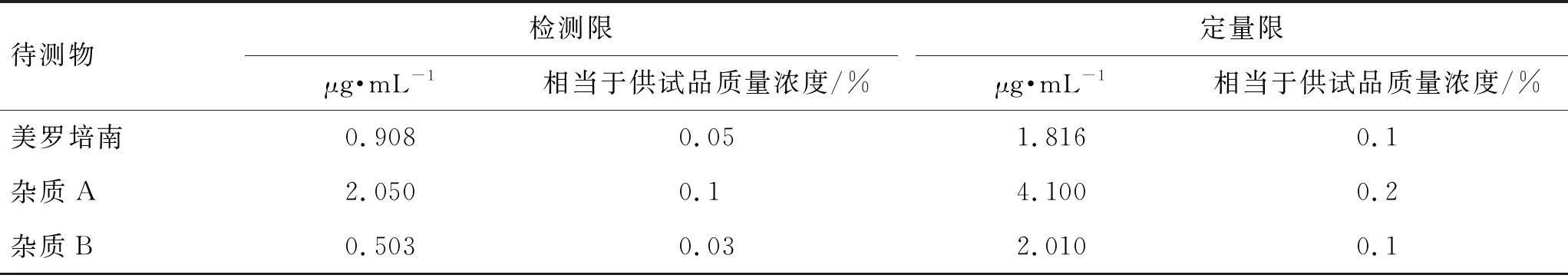
表2 检测限与定量限实验结果
2.6.5定量方式的考察 选取3个企业各1批样品按2.1项下方法制备供试品溶液,并分别精密量取1 mL置于100 mL量瓶中,用水稀释至刻度,摇匀,作为自身对照溶液,进样测定。各批样品分别按峰面积归一化和峰面积自身对照法计算杂质总量百分数。结果,企业A(批号2434C)为0.15%、0.16%;企业D(批号20047911)为0.39%、0.40%;企业H(批号9200301043)为0.26%、0.26%。2种定量方式所得结果一致,考虑到本品溶液状态下的不稳定性,选择归一化法进行定量计算。
2.6.6方法耐用性 使用同一批样品,对流动相pH(6.8、7.2)、流速(0.7、0.9 mL·min-1)、色谱柱温度(25、35 ℃)小范围变化对检测结果的影响进行考察。结果,美罗培南理论板数、与杂质峰分离度、杂质总量检测结果等主要参数无明显变化。
2.7 RP-HPLC法的杂质校正因子的测定
《中国药典》2020年版未规定美罗培南杂质A、杂质B的校正因子,欧洲药典10.0版规定杂质A校正因子1.6,YBH02582015标准规定杂质A校正因子1.62,杂质B校正因子1.98。本文基于2个药典方法,对杂质的校正因子进行确认。
取美罗培南对照品,按2.2项下方法,配制系列溶液;取美罗培南杂质A、杂质B对照品,按2.2项下方法,配制系列溶液,分别进样分析,记录色谱图,以峰面积对其质量浓度进行线性回归,得线性方程,并计算校正因子。
美罗培南:y=6.104 5x-0.108 9,线性范围为0.40~40.38 μg·mL-1,r=1.000 0。
杂质A:y=3.859 0x-0.737 3,线性范围为0.99~39.49 μg·mL-1,r=1.000 0;校正因子为1.582。
杂质B:y=5.257 7x-3.073 4,线性范围为0.38~38.20 μg·mL-1,r=0.999 1;校正因子为1.161。
2.8 样品测定结果的比较
将国内上市的11个企业共33批样品分别按2.1、2.2项下方法测定,比较杂质检测结果,见表3。其中,HPSEC法由于杂质A、杂质B不能分离,未引入校正因子,结果由峰面积归一化法计算;RP-HPLC法中,杂质A校正因子为1.582,与《欧洲药典》中1.6一致,杂质B为1.161,《欧洲药典》中并未规定,考虑到测试误差等因素,杂质B结果没有校正。
《中国药典》2020年版有关物质规定最大单杂质不得过0.5%,总杂质不得过1.5%。RP-HPLC法项下33批产品均符合规定,其中企业D、F、H、I杂质含量较高,接近限度,企业A、C杂质含量明显好于其他厂商,H通过一致性评价后企业的产品杂质水平明显低于本厂其他产品。本品制剂处方简单,而杂质方面仍与原研产品存在明显差距。
国家药品标准YBH00182011、YBH02582015、YBH02162016规定聚合物杂质限度1.0%、0.8%、1.5%,HPSEC法项下所有批次产品均符合规定,且大幅低于限度。HPSEC法总杂质结果明显低于RP-HPLC法,说明本法杂质定量结果准确性较低,可能由于在254 nm波长处检测灵敏度低导致杂质漏检以及杂质A、杂质B没有引入校正因子。

表3 HPSEC和RP-HPLC 2种方法检测杂质结果比较
3 讨论
3.1 HPSEC法的专属性
本文在YBH00182011、YBH02582015、YBH02162016 3个国家药品标准基础上,参考相关文献,建立了HPSEC法对美罗培南在不同降解条件下产生的聚合物杂质进行分析,结合主要降解杂质的光谱信息并与杂质对照品峰位的比对发现,HPSEC法对分离聚合物和小分子杂质的专属性存在问题,这也是目前用凝胶色谱方法对美罗培南聚合物进行分析的共性问题[20]。该问题的解决有待高效凝胶色谱柱填料技术的进一步提高以及多种现代色谱、质谱技术的联合应用[21]。
3.2 HPSEC法的研究现状和建议
在已有文献和标准中,HPSEC法研究美罗培南聚合物通常在254 nm波长处检测[7-9],这对易于水解形成开环的杂质响应很低(见表1杂质A紫外光谱),本研究在此条件下测得杂质A校正因子为6.7,二聚体杂质B校正因子为2.2,而已有文献和标准均未采用校正因子,致使杂质含量被低估。《中国药典》2020年版、《美国药典》2021版和《欧洲药典》10.0版在美罗培南原料或制剂项下均未收载聚合物检查项,而是通过有关物质项控制,通过本文2种方法的对比也表明反相液相色谱法能够对小分子杂质和聚合物杂质同时控制,但方法仍需改进,比如特定杂质的校正因子已经超出0.9~1.1的范围应予引入,使定量更准确。因此,作者认为在国家药品标准[11-13]中采用HPSEC方法控制聚合物的必要性值得商榷。
