基于高通量测序技术的胆囊癌患者肠道菌群变化特点
2023-01-16陈文辉于龙刘文韬翟文龙
陈文辉,于龙,刘文韬,翟文龙
(郑州大学第一附属医院 肝胆胰外科,河南 郑州 450052)
胆囊癌(gallbladder cancer,GBC)是常见的胆道恶性肿瘤,在同期胆道疾病中,GBC的发病率达0.4%~3.8%,在整个消化系统恶性肿瘤中居第六位,且逐年上升,由于GBC发病隐匿,多数确诊时已属中晚期,导致GBC患者病死率高,生存质量极差[1-3]。研究证实,在胆道系统恶性肿瘤各种发病因素中,微生物因素被认为是重要的因素之一[4]。随着高通量测序技术的发展,研究发现有数万亿的微生物寄居在人类胃肠道,其主要致癌机制包括介导炎症反应、改变免疫应答、产生致癌代谢物和DNA损伤[5-6]。肠道菌群在肝外胆管恶性肿瘤中的作用已经得到广泛关注[7]。肠道菌群在胃肠道肿瘤中所起作用已有文献证实,在肝、胰腺等其他消化系统肿瘤中可能的作用也陆续有研究报道[8-9]。目前,关于GBC发病过程中肠道菌群变化特点和潜在机制的研究在国内外鲜有报道。因此本研究采用高通量测序技术,探讨GBC患者肠道菌群分布规律及变化特点,并通过PICRUSt2预测菌群功能差异,旨在阐述GBC人群中肠道菌群特点及其与GBC发生发展的关系,为探索肠道菌群在GBC中的具体机制及疾病的潜在治疗方法奠定基础。
1 材料和方法
1.1 样本收集选取2020年9月至2021年9月在郑州大学第一附属医院肝胆胰外科就诊的14例GBC患者为研究对象(Group_1:N1001~N1014),诊断符合2020年美国国立综合癌症网络肝胆肿瘤临床实践指南(Ⅴ1版)[10],其中男8例,女6例,年龄为(53.20±10.58)岁;并招募9例健康志愿者作为健康对照组(Con:B16S26~B16S34),两组患者性别、年龄相匹配。用一次性无菌收集勺收集清晨粪便至无菌粪便收集器中,置于冰盒送往实验室,分装后立即置于-80 ℃冰箱中进行冻存待检,冻存时间少于1个月。纳入标准:GBC组病理诊断为原发性胆囊恶性肿瘤。排除标准:1个月前使用抗生素或益生菌治疗史;放化疗、靶向免疫治疗史;胆道系统手术史;其他肿瘤史。本研究通过郑州大学第一附属医院医学伦理委员会批准(2018-KY-83),在研究开始前均签署知情同意书。
1.2 16S核糖体RNA(16S rRNA)基因测序使用基因提取试剂盒提取样本基因组DNA,使用琼脂糖凝胶电泳及NanoDrop2000分光光度计检测DNA浓度。为了确保扩增效率和准确性,根据选择的测序区域,使用带barcode的特异引物,Takara公司的Tks Gflex DNA Polymerase,以基因组DNA为模板,进行聚合酶链反应(polymerase chain reaction,PCR)扩增。挑选16S V3~V4区(引物343F和798R)作为细菌多样性鉴定对应区域。采用电泳检测法对PCR产物进行检测,后经纯化、二轮PCR、再检测、再纯化,最后用Qubit定量法对产物进行定量。等量混样后,上机测序。
1.3 微生物分析1个可操作分类单元(operational taxonomic unit,OTU)指相似度大于或等于97%的参数序列。使用QIIME软件包挑选出各个OTU的代表序列,并将所有代表序列与数据库进行比对注释。使用R软件评估不同样本之间的相似性,并根据OUT信息进行聚类分析和主坐标分析(principal co-ordinates analysis,PCoA),绘制PCoA图。利用TAYC系数计算每个样本进化微生物群落之间的距离,表示为两组之间不显著的聚类树。算术平均法用于描述多个样本之间的差异(即1-相似度)。采用线性判别分析(linear discriminant analysis,LDA)效应大小(LDA effect size,LEfSe)方法筛选特异性菌群(默认线性判别值为4)。利用R语言进行关联与模型预测分析。基于PICRUSt2进行差异菌群的京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)[11]代谢途径功能预测。
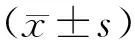
2 结果
2.1 样本OTUs统计及分布通过花瓣图可知,14个GBC组粪便样本与9个健康对照组粪便样本共有39个OTUs,分组后,韦恩图分析可见,在GBC组和健康对照组中有2 320个相同OTUs,除此之外,823个OTUs和1 550个OTUs分别为GBC组和健康对照组各自特有的。见图1。

A为花瓣图;B为韦恩图。图1 OTUs分布图
2.2 GBC组与健康对照组α多样性为了分析生物环境内物种的多样性程度,采用Chao1指数、Shannona指数、goods_coverage指数、observed_species指数、simpson指数5个α多样性指数。Chao1指数越大、Shannona指数越高表明微生物丰富度越大、多样性越高;goods_coverage指数能较为真实地反映样品的测序深度;observed_species指数大小能够很好指示测序数据量的合理性;simpson指数用来表征群落内物种分布的多样性和均匀度,5个α多样性指标在两组样本间均为GBC组低于对照组,差异有统计学意义(P<0.05)。见表1。

表1 两组细菌间多样性比较
2.3 GBC组与健康对照组患者β多样性采用非加权和加权Unifra的2种PCoA法分析GBC组与健康对照组的微生物群落结构差异,发现两组的肠道细菌群落存在显著差异,各自分布在相对独立区域。见图2。

A为非加权PCoA图;B为加权PCoA图。图2 β多样性分析PCoA图
2.4 不同水平肠道微生物区系的丰度和结构变化在所有样本检出的菌门中,前4位优势菌门分别为Proteobacteria、Firmicutes、Bacteroidetes、Actinobacteria,GBC组和健康对照组相同。与健康对照组相比,GBC组Proteobacteria相对丰度增高(t=4.265,P<0.001),Bacteroidetes相对丰度减少(t=6.533,P<0.001)。较健康对照组,GBC组Firmicutes相对丰度有增加的趋势,Actinobacteria相对丰度有降低趋势,但差异无统计学意义(t=0.359、1.000,P>0.05)。GBC组与健康对照组相比,在门水平上,7个菌门差异有统计学意义(P<0.05),分别为Bacteroidetes、Proteobacteria、Entotheonellaeota、Acidobacteria、Patescibacteria、Dependentiae、Deferribacteres(t分别为6.533、4.265、3.133、3.031、2.465、2.352、2.191,P<0.05)。GBC组和健康对照组具有统计学差异的细菌在属水平上共有140种,其中前10菌属分别为Escherichia-Shigella、Bacteroides、Prevotella_9、Prevotella_2、Parabacteroides、Parasutterella、Alistipes、Lachnoclostridium、Lachnospiraceae_NK4A136_group、Lachnospira(P<0.05)。见图3。

A为门水平相对丰度分布图;B为门水平前4位菌门直方图;C为属水平前10位菌属分布箱式图;D为属水平前15位差异菌群分布热图;***表示P<0.001。图3 门、属水平菌群分布图
2.5 LefSe分析筛选差异菌属LefSe分析揭示两组或以上生物群落差异物种的组成差异。物种注释分支例图展示GBC组和健康对照组在不同分类水平上的最大不同。根据LEfSe分析结果,在GBC组中,Enterobacteriales、Proteobacteria、Klebsiella、Lactobacillales丰度高于健康对照组;Roseburia、Prevotella_2、Prevotella_9、Bacteroidaceae丰度低于健康对照组,差异有统计学意义(LDA=4,P<0.05)。见图4。

A为差异物种注释分支例图;B为差异物种score图。图4 LefSe分析图
2.6 关联和模型预测分析以丰度前30的属为对象,利用R包绘制相关性heatmap图,进行Indicator分析和随机森林图绘制,结果如下。见图5。

A为相关性heatmap图,红色为正相关,蓝色为负相关;B为Indicator指示性物种范例图;C为重要物种点图,Mean Decrease Gini值越大表示该变量的重要性越大。图5 关联和模型预测分析图
2.7 基于16S的KEGG功能预测利用PICRUSt2预测GBC组与健康对照组之间的KEGG通路,选取差异最显著的前30条代谢通路:糖胺聚糖降解、溶酶体表达、脂肪细胞因子信号通路等代谢通路表达减弱(P<0.05);次生代谢产物的生物合成与降解、不饱和脂肪的生物合成、电子转移载体等代谢通路表达增强(P<0.05)。见图6。

图6 KEGG差异结果聚类heatmap图
3 讨论
肠道微生物区系参与物质和能量的新陈代谢,促进免疫系统的发育和成熟,形成黏膜屏障,避免病原体攻击[12]。通常,肠道微生态的平衡需要肠道菌群和宿主共同维持[13]。多种慢性疾病如结肠炎、结肠癌、胆结石[14]以及肥胖和糖尿病等代谢性疾病都是由于这种平衡被打破导致的[15]。有研究证实,其他消化道肿瘤如胃癌、肝癌等及神经精神疾病均伴随肠道菌群失调[8,16-17],说明肠道菌群可能还有更多未知作用需要探索。就肝外胆道系统而言,国内外研究相对匮乏。李涛等[7]通过收集肝外胆管癌、胆管结石和正常人群粪便标本证实3组肠道菌群存在差异,由此推断肠道菌群失调在疾病发展中可能发挥着重要作用。基于以上研究,本研究欲探讨GBC患者肠道菌群是否存在一定联系。
本研究结果显示,在α多样性分析中,与健康对照组相比,GBC患者肠道菌群的Chao1指数和Shannon多样性均降低。Sarhadi等[18]在关于胃癌的一项研究中取得了类似的结果。本研究结果与Wheatley等[19]发现远端胆管癌患者肠道菌群多样性高于正常健康人不同,后续需要更深入研究。就α多样性而言,与Chao1指数相比,Shannon多样性在描述微生物群落丰富度和均匀性方面更有优势。因此,肠道菌群Shannon多样性下降可能标志着GBC患者疾病状态。GBC患者肠道菌群与正常健康组相比存在一定差异,在人体四大菌门中,Proteobacteria在GBC组中较健康对照组相对丰度增加。Proteobacteria中多种条件致病菌能产生脂多糖,脂多糖是一种内毒素,是引起炎症反应的重要物质。研究表明,长时间暴露于脂多糖的大鼠可导致肝快速损伤[20]。根据LefSe所示,Enterobacteriales、Proteobacteria、Klebsiella、Lactobacillales在GBC组中相对丰度增加。Escherichia-Shigella在GBC患者肠道菌群中富集,Bacteroides、Parasutterella等在GBC患者肠道菌群中的丰度有所降低。Escherichia-Shigella是肠杆菌主要菌属之一,是人体重要致病菌之一,正常情况下在肠道内与其他细菌处于平衡状态。Escherichia-Shigella在人体内主要有两方面重要作用,其一,Escherichia-Shigella在诱发肠道炎症方面扮演重要角色;其二,在炎症环境中,Escherichia-Shigella可通过分泌肠杆素获得更强的生存优势,加剧肠道微生态的紊乱[21]。根据Proteobacteria和Escherichia-Shigella研究结果,推测炎症反应在GBC疾病演变中发挥重要作用。有研究报道,肠杆菌科的富集程度可能与某些肿瘤分期有关,如胃癌,且肠杆菌科在胃癌患者肠道中富集意味着患者的死亡风险更高[22]。Bacteroides和Parabacteroides均为益生菌。Bacteroides可产生维生素K并具有抗炎能力[23];而Parabacteroides除营养、免疫增强等作用之外,还有一定的抗肿瘤作用[24]。二者丰度降低可能在GBC的发生发展过程中发挥一定作用,具体机制有待进一步研究。在相关性和功能预测分析模型中,Escherichia-Shigella可定义为在GBC一定区域范围内能对生长环境产生较大影响的菌属,Parasutterella作为区分GBC组和健康对照组间差异的关键性物种,其促进MCA-205肉瘤的作用在体外试验中已有报道[25],是否在GBC发病过程中发挥重要作用和能否作为GBC新的早期诊断标志菌群尚未可知。KEGG功能预测展示出差异菌群在代谢途径上的潜在作用,但其具体作用机制仍未可知,需要进一步更深层次的研究。
综上所述,本研究发现肠道菌群在GBC患者和健康人群的变化特点,表明肠道微生物在GBC发生发展过程中发生连续性变化,但本研究存在一定局限性,如样本量较小、单中心研究等。未来需要大样本量、多中心、多组学进一步探讨GBC发生发展的分子机制与肠道菌群变化的内在联系,不仅有利于加深对GBC发病机制的理解,还有助于探寻GBC早期诊断标志物和探索开发新型微生物疗法。
