离子液体功能化金属/共价-有机框架材料在催化反应中应用
2023-01-16兰永成鲁艳梅夏春谷刘建华
兰永成, 鲁艳梅, 郧 栋, 夏春谷, 刘建华*
(1. 中国科学院兰州化学物理研究所 羰基合成与选择氧化国家重点实验室, 甘肃 兰州 730000;2. 中国科学院大学, 北京 100049)
金属有机骨架材料(Metal organic frameworks,MOFs)作为一类由无机金属和有机连接体通过桥联配位作用形成的具有代表性的永久多孔框架结构材料, 通常有着比表面积大、 孔径和孔道结构可调、 金属中心与配体丰富多样以及易于功能化等特点[1-2]. 作为继MOFs之后的又一重要多孔材料, 共价有机框架材料(Covalent organic frameworks, COFs)是一类由复杂的有机结构通过可逆共价键(C=C、C=N、 C-N、 B-O)连接而成的二维或三维多孔结晶材料. 除了与MOFs相似的比表面积大、 结晶度高和开孔结构特性以外, 高的热化学稳定性、 低的骨架密度以及合成策略多样化成为COFs材料的独特优势[3-5]. 到目前为止, MOFs和COFs材料已经在气体吸附分离、 生物及化学传感、 储能材料、 光电功能材料和催化化学等领域展示出广阔的应用前景. 基于MOFs和COFs材料的特殊物理和化学性质,特别是可以利用构筑基元的变化来调变MOFs和COFs材料的结构、 组成及功能, 其基元单元具有易于修饰改性和功能化的优点. 在催化化学研究领域,MOFs和COFs材料主要是作为催化活性中心载体材料或者利用骨架中含有催化功能的有机片段来发挥作用.
MOFs材料在催化领域的应用主要分为3种不同的类型: 1) 金属节点在催化反应中起主导作用,然而, 由于金属中心易受到有机溶剂占据或有机配体部分饱和的不利影响, 导致金属位点或金属簇作为催化活性位点的研究并不多见; 2) 有机配体组分发挥催化性能, 目前来说, 能够用于合成MOFs的有机配体主要是含氮的芳香族分子或是羧酸盐衍生物, 种类较为有限, 因此MOFs中具有催化活性的有机配体的研究也屈指可数; 3) MOFs作为一种多功能载体材料负载具有催化活性的组分起主要催化作用, 由于MOFs作为载体提供天然的物理空间进而应用于各类有机催化反应当中, 因此成为MOFs在催化领域应用研究的重点[6-7].
COFs材料在催化领域的应用主要分为两种不同的类型: 1) 充分利用构筑COFs材料的有机单元酸碱性的特点, 特别是利用富含氮元素的COFs材料本身具有丰富的碱性活性催化位点, 来催化Knoevenagel缩合、 加成等有机反应; 2) COFs作为载体材料, 与各式各样的活性位点(如单金属位点、 金属纳米颗粒和金属氧化物等)相互组合得到COFs负载型的多相催化材料, 进而在碳-碳偶联反应、 CO2转化、 光电催化等反应中得到应用[8-9].
然而, 目前MOFs和COFs材料在催化反应中的应用还比较有限. 为了丰富拓展MOFs和COFs在催化领域中的应用范围, 将离子液体(Ionic liquids, ILs)与MOFs或COFs有效耦合, 制备出新型功能化材料不失是一种有益的选择. 离子液体是一类由有机阳离子与有机/无机阴离子构成的创新型材料, 凭借着其具有良好的热/化学稳定性、 极低的蒸气压、 结构可设计等特性, 已在包括化学、 能源、 环境、 材料等众多的领域得到广泛的应用[10-13]. 将结构和功能可调控的离子液体和多孔MOFs和COFs组合形成结构、 功能多变、 种类多样的离子液体功能化的MOFs和COFs新材料, 既保留了MOFs和COFs的高比表面积、 丰富的表面性质、 有序结构、 可调节的孔隙功能和孔结构的优点, 又兼具离子液体独特的结构和热稳定性高等优势, 同时新材料还具有离子界面, 可以与相反电荷的物质产生强相互作用, 能伴随新的作用位点和特性的产生. 因此, 离子液体功能化的MOFs和COFs新材料是一种极具潜力的催化材料.
制备离子液体功能化的MOFs和COFs材料的主要策略包括: 物理浸渍或自下而上、 后修饰等[14].物理浸渍法是离子液体功能化的MOFs和COFs材料中最直接的方法, 利用离子液体与MOFs和COFs材料物理混合, 然后再经过加热处理, 使得离子液体充分扩散到MOFs和COFs孔内. 自下而上策略是将离子液体功能化单元预先嫁接到MOFs或COFs上, 再通过单体的进一步转化得到具有本征型离子液体功能化的多孔杂化材料. 这种离子液体功能化MOFs和COFs的制备方法优势在于赋予骨架强大功能性、 调节孔道内的微环境以及活性位点的均匀分布, 同时还能够通过对单体的预先设计调整杂化材料的孔道形状和孔径大小. 后修饰的方法主要有3种: 1) 通过在MOFs和COFs材料的有机骨架中引入活性官能团(-OH、 -CH3Cl、 -NH2)作为锚定位点, 充分利用离子液体单元和锚定位点的化学作用, 以共价键连接方式将离子液体片段引入到MOFs和COFs材料骨架结构中; 2) 通过分子间作用力或氢键等非共价键作用, 即将带有乙烯基离子液体的单体材料连接到MOFs或COFs中, 并在偶氮二异丁腈(AIBN)为引发剂条件下通过自由基聚合过程得到离子液体功能化的MOFs和COFs材料; 3) 通过离子化的方法,即利用无机酸或无机盐离子化试剂与中性的MOFs或COFs发生相互作用构筑离子型杂化材料.
毫无疑问, 这种离子液体功能化的MOFs和COFs杂化材料不仅能“扬长避短”, 还兼具以下独特优势: 1) 离子液体片段掺杂可以进一步丰富MOFs和COFs框架材料多样性, 有助于拓展其在催化化学中更加广泛的应用; 2) 二者兼具灵活的骨架结构, 有利于MOFs或COFs与离子液体片段的相互作用, 从而发挥出双重或者多重优势达到优异的催化性能. 在离子液体功能化的MOFs和COFs材料中,MOFs和COFs材料与生俱来的丰富孔道结构与超高比表面积为负载均匀分散的催化活性中心提供了可能, 同时使催化剂与反应底物充分接触, 促进底物与产物的传质过程, 进而实现催化反应的高效进行.离子液体片段的引入, 可以作为催化活性中心的配体(稳定剂)或分散剂, 同时能够有效改善MOFs和COFs材料孔道和活性中心周围的微环境; 另外, 还可以充分利用离子液体片段在适当的反应条件下转化为氮杂环卡宾配体的特点, 在MOFs和COFs材料中引入氮杂环卡宾有机金属配合物.
目前离子液体修饰的MOFs和COFs框架材料主要应用在气体储存、 吸附和基于膜的气体分离、离子导电等领域, 并且显示出优越的性能, 尽管在部分催化过程中已经有相关的零散报道[15-16], 但是缺乏对离子液体功能化的MOFs和COFs在各类有机催化反应中应用进展的系统性介绍. 围绕离子液体修饰的框架材料在催化反应中的研究现状, 我们总结了近几年来离子液体功能化的MOFs和COFs材料用于催化CO2环加成、 CO2还原功能化应用、C-C偶联反应及羰基化反应等其它相关催化反应,并简要探讨相关催化反应机理, 同时对该领域的局限性和发展前景进行总结和展望.
1 CO2环加成反应
最近几十年里, 汽车尾气排放、 化石燃料燃烧等人类活动导致温室气体CO2排放量急剧增长, 由此所带来的全球变暖和气候变化等环境问题成为当今社会的一大难题[17]. 然而, 从另一方面来讲, CO2作为未来最丰富的碳源之一, 使用这种无毒且廉价的化学原料并将其转化为高价值的化学品将是一种消除温室气体CO2的高效途径. 事实上, 随着科学技术的进步, 大量的研究努力已经实现了将CO2与环氧化物反应制备环状碳酸酯(图1). 此外, 环状碳酸酯普遍用作极性非质子溶剂、 锂离子电池电解质溶液和有机中间体等[18-19], 在实际应用中具有重要意义.
然而, CO2本身具有较高的热力学稳定性和动力学惰性, 严重限制了在有机合成中的利用, 因此催化剂成为其催化转化的关键因素. 离子液体因具有独特的性能(蒸气压低、 优异的热和化学稳定性),在作为催化剂方面已取得重大进展, 并且也有研究证明离子液体具有强亲CO2性能, 能够有效促进CO2固定与转化[20]. 例如中科院化学所韩布兴课题组[21]发展了离子液体与银盐AgCl/[Bmim][OAc]催化体系, 对CO2和炔醇合成不对称有机碳酸盐表现出较高催化性能. 然而, 目前均相体系在CO2固定反应中仍然存在局限性, 例如离子液体大量使用导致反应过程中底物分子传质阻力增大, 进而影响活性与选择性, 此外, 反应条件苛刻与分离纯化成本高也不容忽略. 为了解决上述问题, 有学者提出将离子液体固载到MOFs和COFs上, 将离子液体的溶解性特点与载体材料的高比表面积优点融会贯通,能够有效改善均相催化体系的缺点, 同时保持优异的催化性能. 但利用MOFs和COFs催化CO2环加成反应往往需要高压条件或者加入大量的有机碱, 这不仅增大了成本和危险性, 而且有机碱会堵住孔道,严重阻碍催化剂的循环使用.
在此启发下, 研究者利用合理的离子液体与金属有机MOFs或共价有机COFs框架材料相结合, 构建离子液体功能化的多孔催化材料. 一方面, 该材料具有MOFs和COFs的拓扑构型, 同时催化剂中咪唑离子液体的存在能够增强对CO2的吸附, 其中酸碱位点与亲核试剂的作用可以实现在无溶剂常压条件下协同催化环氧化物与CO2生成环状碳酸酯; 另一方面, 离子液体功能化MOFs或COFs材料保持催化活性基本不变, 同时使离子液体异相化, 最大程度地达到了“一石二鸟”的作用[22].
总体上来说, CO2与环氧化物的环加成反应生成环状碳酸酯通常遵循环氧化合物活化与CO2活化两条路径, 环氧化物活化过程中主要是亲核试剂攻击环氧化物上位阻小的碳原子; 而CO2活化过程则是亲核试剂攻击CO2上的亲电碳原子. 对于在此探讨的离子液体功能化MOFs和COFs材料催化CO2环加成反应普遍遵循环氧化物活化机理[23-25], 包括如下步骤(图2): (1) 通过离子液体功能化框架材料的Lewis或Brφnsted酸位点与环氧化物中弱碱性氧原子相互作用激活环氧化物. 在含有金属的催化剂中,相互作用主要是通过金属氧配位键, 而环氧化物也可提供氧原子与含羟基、 羧基或离子液体中2位质子的杂化材料形成氢键从而被激活; (2) 接着, 卤化物阴离子(X-)亲核进攻环氧化物中阻碍最小的碳原子引起环氧化物的开环形成醇氧化物, 而MOFs或COFs自身有序的骨架结构能够稳定环开的醇氧化物中间体; (3) 随后, 通过离子液体静电作用或具电负性的氧作为亲核试剂攻击亲电的碳原子激活CO2,形成碳酸盐化合物; (4) 开链的碳酸盐发生分子内环消去反应; (5) 最后, 卤素阴离子置换生成五元环碳酸酯产物, 催化剂再生用于下一次循环. 由此看来,离子液体功能化MOFs或COFs框架材料在环加成反应中影响巨大, 加速了环氧化物的开环进程、 确保醇氧中间体的稳定化以及活化CO2都与其密切相关.

图2 利用IL/MOFs(COFs)合成环状碳酸酯一般机理[23-24]Fig.2 General mechanism of cyclic carbonate synthesis using IL/MOFs(COFs) [23-24]
2017年, Ding等[26]通过将咪唑离子液体直接配体功能化的方法合成出的UiO-67-IL杂化材料用于催化环氧氯丙烷(ECH)与CO2环加成反应, 在不存在共催化剂四丁基溴化铵(TBAB)时, 0.1 MPa, 8 h,90 ℃可高达95%的环状碳酸酯产率, 而添加TBAB后, 能够大幅度缩短反应时间并接近完全转化. 此外, 催化剂循环5次后产率仍然保持在96%以上. 随后, 他们[27]采用类似方法用咪唑修饰的有机配体制备出一系列UiO-66-IL纳米颗粒(NPs), 再利用NPs上的OH-基团与聚氨酯低聚物化学交联构建出离子液体修饰的MOF-聚合物薄膜(UiO-66-IL-ClO-4). 在常压条件下, 添加TBAB催化ECH环加成反应, 产率高达99%.
2018 年, Ji课题组[28]通过简单通用的串联后合成修饰法将离子液体固定在MIL-101-NH2上并成功制备了一种新型酸碱双功能化催化剂(IL/MIL-101-NH2), 并用来评价以CO2和环氧丙烷(PO)为原料的环加成反应的性能, 由于酸碱位之间的协同作用, 即使不使用任何助催化剂, IL/MIL-101-NH2多相催化剂对CO2与PO反应生成环状碳酸酯仍具有高效的催化效率. 在1.3 MPa的低压环境下和1 h的极短的反应时间下, 环状碳酸丙烯酯的产率可达91%.值得注意的是, 催化剂循环使用5次后, 没有显著的催化活性损失.
Park团队[29]先合成出MOF的一个亚类n-ZIF-90, 然后首次将氨基吡啶碘化物离子液体(AmPyI)同化到n-ZIF-90 材料上得到新颖的单组分催化剂IL-ZIF-90. 作者采用XRD对上述两种材料进行表征可以看出, 合成的n-ZIF-90与模拟的ZIF-90的峰大致吻合, 并且峰强度的变化也足以说明离子液体的成功引入. 为了证明其催化性能, 作者选择在不存在助催化剂与溶剂的情况下, 催化环氧化合物与CO2制备环状碳酸酯. 选用PO作为底物, 在120 ℃、1 MPa条件下反应3 h有着明显的催化效果(95%产率与98%选择性). 接着, 在同样的反应条件下考察了IL-ZIF-90对不同底物的活性, 他们发现该催化剂对各种末端环氧化物(EPH、 AGE、 SO)都具有优异的反应效果. 此外, IL-ZIF-90催化剂具有优异的稳定性, 在连续重复使用4次循环后, 催化活性没有明显的损失, 选择性仍保持在98%.
根据以往的研究发现, 含有COOH-基团的咪唑IL由于氢键的作用, 在CO2环加成反应中具有优异的催化性能. 2019年, Bahadori等[30]及其同事先后通过氯甲基化、 嫁接咪唑和季胺化反应制备出任务特异性的多相催化剂MIL-101(Cr)-TSIL. 在110 ℃,2 h, 2 MPa下催化CO2与氧化苯乙烯(SO)可高达98%转化率和97%选择性, 该催化剂在重复利用5个循环后依然保持较高的转化率与选择性, 表明催化剂具有较高的稳定性. 此外, 在相同条件下, 可以催化各种环氧化物得到高收率、 高选择性的相应环状碳酸酯产物. 这很大程度是因为COOH-、 咪唑和MIL-101三者的协同作用产生的效果, 环氧化物与COOH-作用产生氢键, 接着Br-以Lewis碱的形式亲核进攻环氧化物空间位阻小的碳原子, 生成开环的环氧化中间体, 紧接着咪唑盐阳离子由于静电作用导致二氧化碳分子活化并插入, 最后环状碳酸酯产物生成; 另一方面MOF材料的介孔结构提高了IL周围CO2浓度, 从而三者的协同效应大大提高了催化效率.
2020年, Bahadori等[31]采用将咪唑基离子液体通过配位键和共价键的形式与MIL-101(Cr)中Cr中心和有机单元分别作用形成MIL-IL(A)和MIL-IL(B)两种多相催化剂(图3), 并在无助催化剂和无溶剂条件下被用于与环氧化物的CO2固定. 用SO作为环加成反应的底物, 在反应温度为110 ℃, 反应压力为2 MPa下反应6 h两种催化剂催化的产率分别为98%和93%. 此外, MIL-101(Cr)B能够连续5次重复使用,转化率只有轻微的下降, 而相比之下, MIL-101(Cr)A在第2次循环中就没有表现出催化活性.
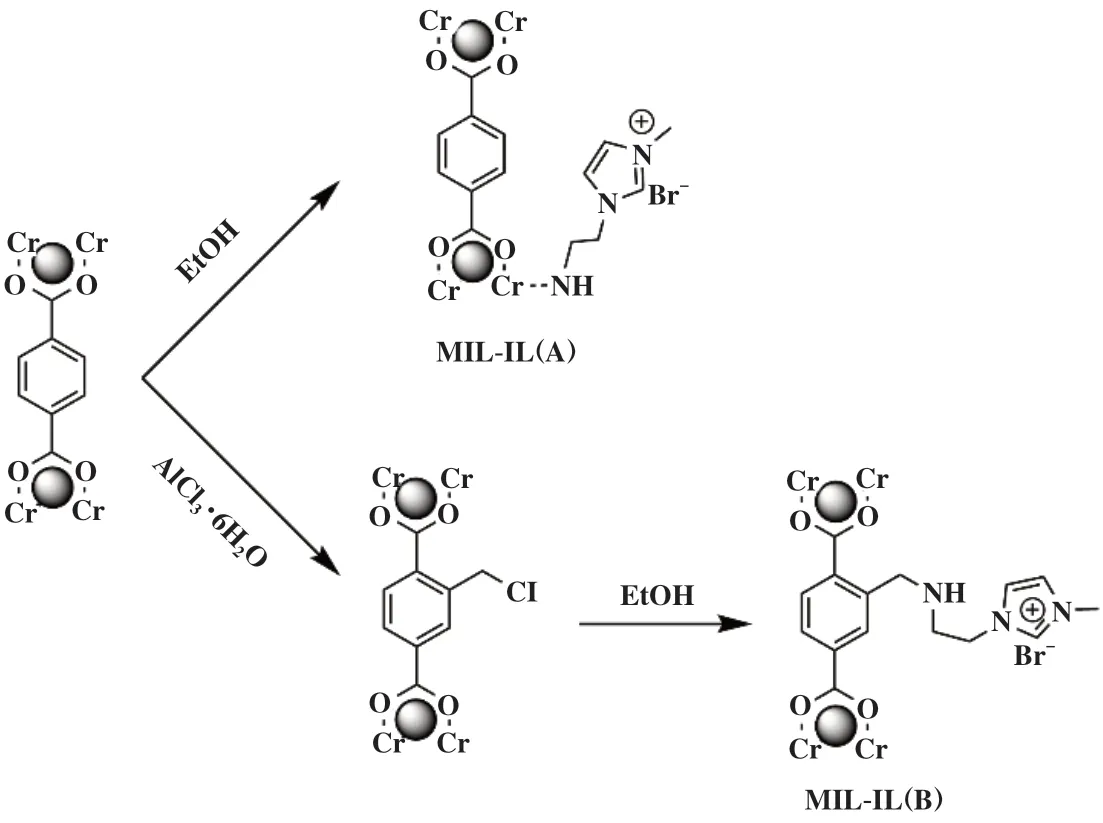
图3 MIL-IL(A)和MIL-IL(B)的合成过程[31]Fig.3 Synthesis process of MIL-IL(A) and MIL-IL(B) [31]
然而到目前为止, 多数报道的催化CO2环加成反应存在着反应条件苛刻(温度高、 压力大、 时间长), 同时无助催化剂前提下表现出较差的催化性能, 这无疑是增加了反应过程的成本. 为了解决以上的问题, 2018年, Liu等[32]采用后合成修饰方法将咪唑IL接枝到MIL-101载体上, 接着利用不同碳链长度的卤代烃烷基化反应合成MIL-101-IMBr催化PO与CO2环加成反应. 他们发现80 ℃, 0.8 MPa,4 h条件下, 含有6 个碳的烷基的催化剂MIL-101-IMBr-6对PO具有最高的催化性能(95.8%转化率和97.6%选择性). 并且在没有严重损失的前提下催化剂能催化循环5次. 无催化剂仍能保持高催化活性主要归因于MIL-101中Cr3+提供路易斯酸位点与PO相互作用.
同年, Ding等[33]报道了用原位封装策略将咪唑基聚离子液体(polyILs)固定在金属有机骨架材料MIL-101中, 所合成的杂化材料polyILs@MIL-101能够有效地催化CO2与环氧化物反应. 该催化剂在温和的条件下(0.1 MPa CO2, 70 ℃), 无需助催化剂对大部分的环氧化物的CO2环加成具有较高的活性. 为了查验其可回收性, 经过离心分离、 洗涤和烘干程序后继续进行后续的CO2环加成反应时发现,polyILs@MIL-101多相催化剂能够持续使用10次而无明显的活性损失.
最近, Jiang 等[34]利用自由基聚合的方式合成咪唑基聚离子液体, 并在MIL-101 框架内进一步原位聚合得到聚离子液体杂化材料. 其中, PIL-4@MIL-101-0.5 催化剂中不饱和配位的Lewis酸性Cr位点与亲核Br-的相互协同作用, 在催化CO2与ECH的环加成反应中表现出良好的催化性能, 没有额外的助催化剂与优化的反应条件下(90 ℃, 1 MPa, 2.5 h)转化率高达99.6%. 此外, 杂化材料具有较好的分离性能和回收稳定性, 至少可以重复使用5次, 同时保持有效的催化活性. 随后, 在以上工作的基础上, Jiang等[35]将氢键供体功能化的聚离子液体引入到MIL-101 框架中, 合成了PIL-R@MIL-101(R=COOH,OH,NH2)杂化材料. 羟基与羧基等氢键供体被普遍认为是环氧化物活化的有效活性位点, 有利于CO2与环氧化物的转化. 因此, 通过Lewis酸Cr中心、 羧基和亲核Br-共同作用对CO2产生良好的亲和力, 使得PIL-COOH@MIL-101催化剂在环氧化物的环加成反应中表现出优异的催化活性.
通过在COFs通道内对各种离子液体进行空间限制与修饰, 可催生出具有靶向性的离子液体功能化COFs杂化材料, 并且能够显著提高催化性能. 目前, 离子液体修饰的COFs材料作为催化剂在CO2固定反应中备受青睐. 2019年, Qiu等[36]报道了采用溶剂热方法合成的咪唑盐功能化的共价有机骨架材料COF-HNU, 其中COF-HNU3表现出较高的BET表面积(2027 m2·g-1), 并且在优化的反应条件下催化二氧化碳与PO环加成反应中可以使产物收率达到99%. 值得注意的是, 该催化剂的转化数(TON)高达495 000. 远超已报道的多相催化剂. 此外, 该催化剂可重复使用至少10次, 且催化活性没有下降. 在每次循环利用后, COF-HNU3的高结晶度和孔隙率基本保持不变. 随后, Zhao等[37]通过酸碱中和作用将IL接枝到COF框架中, 制备出含大量酸性位点的COF材料(TpPa-SO3H),并用水做溶剂将SO3H锚定的COF浸泡在含NH2-的IL无水溶液中, 得到COFHNU14(图4). 在优化的反应条件下(2 MPa, 120 ℃),该催化剂对不同取代基的环氧化物与CO2反应生成相应的环状碳酸酯, 产率高达95% ~ 99%. 此外作者还运用DFT计算研究其机理, 对N-H..Br与CH..Br分子间距离比较发现, 氨基更容易与环氧化物中的O原子形成氢键, 起到活化环氧化物作用, 进而提高催化活性.

图4 COF-HNU14的合成[36]Fig.4 Synthesis of the COF-HNU14[36]
最近, Zhang 等[38]用后合成修饰的方式通过席夫碱缩合反应(Schiff Base Reaction)合成具有羟基的亚胺连接的TD-COF, 然后用[BMIm]Br对OH-进行改性处理, 得到离子液体修饰的COF材料(ImTDCOF). 最后通过静电作用将带负电荷的多金属氧酸盐POM与正电荷的咪唑盐结合制备出多相催化剂(POM@-ImTD-COF). 在TBAB协助催化下, 反应压力为0.1 MPa, 反应温度80 ℃时, 催化SO的转化率高达98%. 此外, 该催化剂对不同底物的环氧化物环加成反应具有普遍适用性, 都表现出优异的催化性能. 出色的性能归因于IL、 POM以及COF材料的共同作用的结果.
2021年, Ding等[39]报道了通过多组分一锅聚合的途径将卟啉与咪唑离子液体官能团引入共价有机框架COF材料中合成COF-PI-1, 随后, 在DMF为溶剂的反应体系中混合Co(OAc)2, 将Co(II)离子对COF-PI-1 进行合成后金属化制备出COF-PI-2 多相催化剂(图5). 由于COF-PI-2 同时存在Co(II)-卟啉与咪唑IL的缘故, 使得其在催化环氧溴丙烷(EBH)与CO2环加成反应中具有高效的反应活性. 值得注意的是, 在可见光照射下催化该反应表现出相同的催化活性, 产物收率也高达99%, 而且COF-PI-2也被证明是一种可循环利用的高效多相催化剂, 经历过5次催化循环后, 环状碳酸酯的产率依旧高达97.5%. 此外, 作者在优化的条件下对不同环氧化物作为底物的环加成反应进行评估, 大部分都能表现出良好的催化活性(>90%).
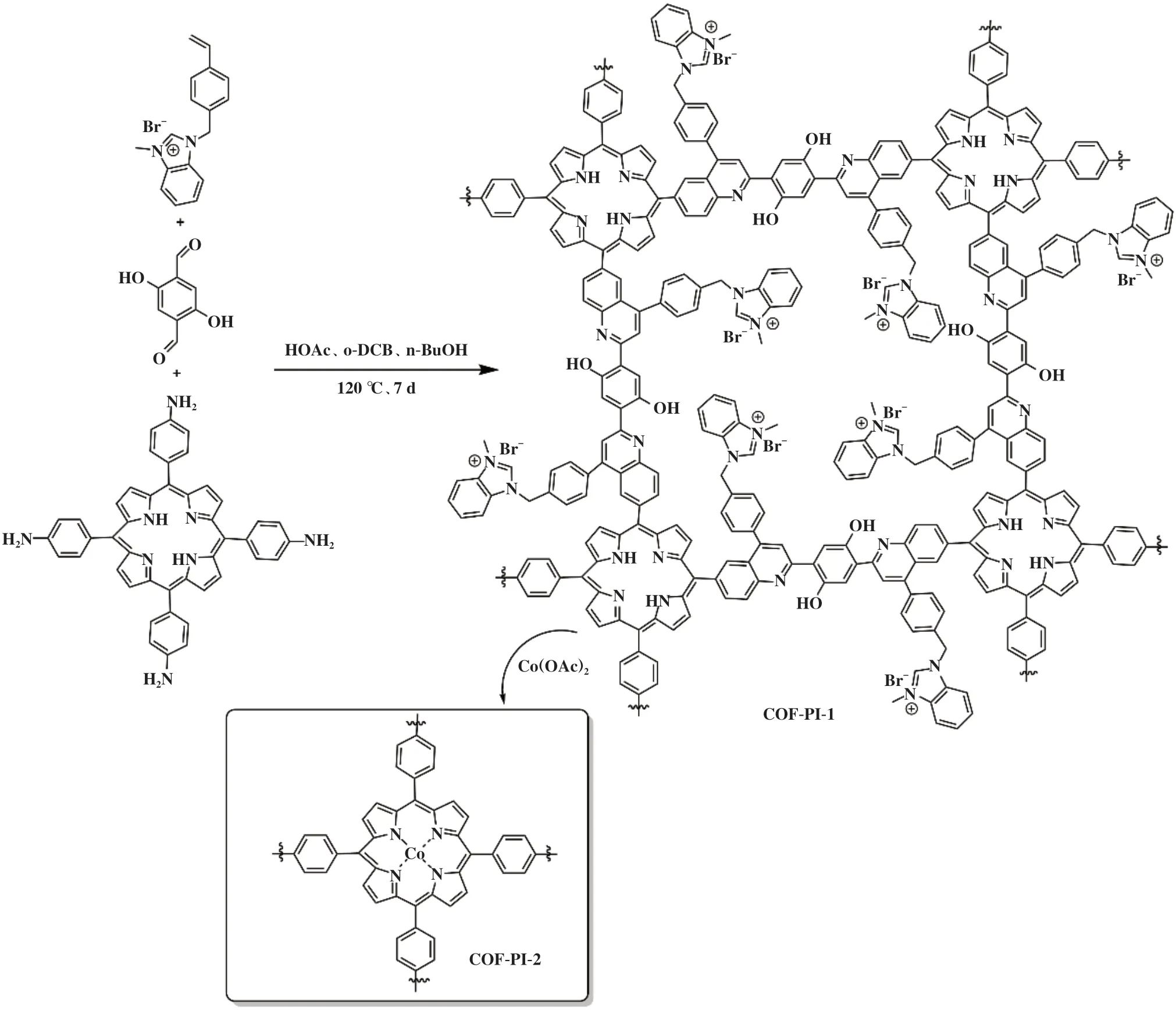
图5 COF-PI-2的合成[39]Fig.5 Synthesis of the COF-PI-2[39]
除了咪唑基团, 富N的三嗪基团也能够作为Lewis碱活化CO2并用于催化环氧化物的环加成反应. 2019年, Biswas及其同事[40]通过自由基聚合方式以AIBN作为自由基引发剂将双阳离子咪唑基IL与三聚硫氰酸TTCA反应制备出聚合催化剂, 利用FT-IR、 solid-state13C CP-MAS等表征手段证明成功聚合. 该催化剂使用KI作为共催化剂, 考察了环境二氧化碳压力下可以有效地催化环氧化物转化为相应的环状碳酸酯, 并且催化剂能够保持其活性长达6个循环. 此外, 他们认为三嗪基团的掺入更有利于吸收CO2并实现咪唑最佳分离, 从而产生更优异的催化性能.
2021 年, Liu等[41]以3-(氰乙基)-1-(4-氰苯基)咪唑氯([CmpIm][Cl])作为单体, 通过离子热聚合也构建出三嗪基共价有机骨架材料; 通过对比发现, 400 ℃及ZnCl2与单体的摩尔比为15∶1时合成的催化剂ImTPOF-400-15(图6)具有更佳的催化活性. 由于具有Lewis酸性的CO2能够分别与咪唑和三嗪基团发生偶极-四重作用和Lewis酸碱作用的影响[42-43], 在存在TBAB共催化剂时能在温和的条件下有效地促进CO2与环氧化物反应生成所需的环状碳酸酯, 此外, 在无明显催化活性降低情况下, 该催化剂可以重复使用5次循环.

图6 ImTPOF-400-15的合成[41]Fig.6 Synthesis of the ImTPOF-400-15[41]
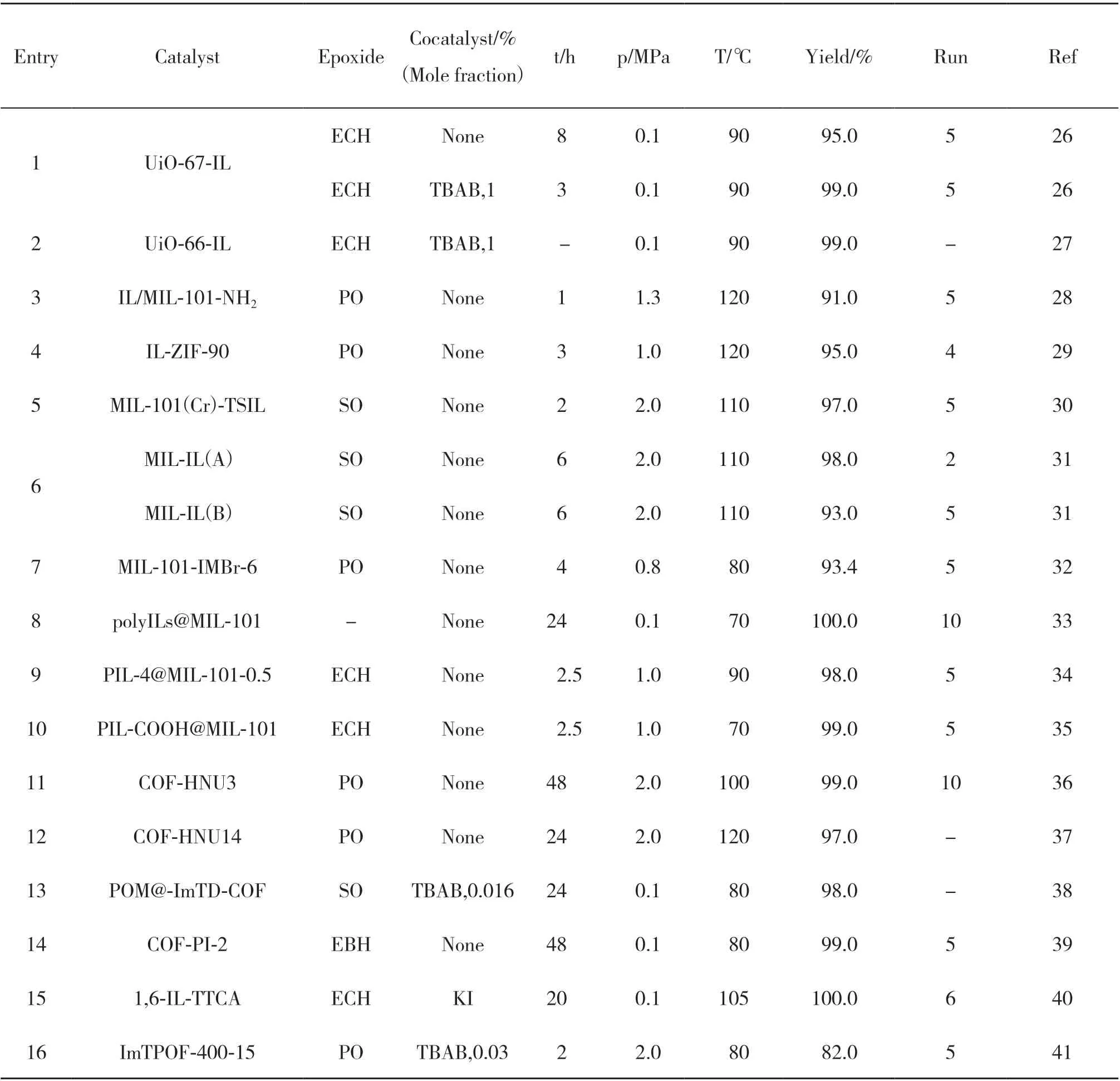
表1 含IL的MOFs和COFs催化体系催化CO2与环氧化物环加成反应Table 1 Cycloaddition of CO2 with epoxides catalyzed by IL-containing MOFs and COFs catalytic systems
2 CO2还原功能化应用
CO2作为一种低成本、 安全、 可再生的碳资源,利用CO2作为原料合成具有高价值能源产品和精细化学品已经成为减少碳排放的一项重要措施. 随着催化技术的飞速发展, 除了在前面提到的CO2环加成反应以外, 利用CO2化学还原转化为甲酸、 羧酸等途径都已被开发出来. 在本节中, 我们主要介绍几种离子液体固载化的框架材料用于CO2加氢制甲酸、 CO2羧化以及CO2硅氢化反应.
2016 年, Dong 等[44]预 先 合 成 出2D COFs 材料, 再利用威廉姆逊醚合成反应(Williamson ether reaction)首次将具与催化活性的离子液体修饰在COFs上制备出无金属的[Et4NBr]x%-Py-COFs复合物材料(图7). 作者将其用于CO2吸附实验发现有着较高的CO2吸附能力和Qst值, 除此之外, 多相催化剂在温和的条件下催化不同胺的甲酰化反应也展现了出色的催化性能(88% ~ 94%), 并且能通过简单的方法分离回收并重复使用4次.

图7 [Et4NBr]x%-Py-COFs的合成[44]Fig.7 Synthesis of the [Et4NBr]x%-Py-COFs[44]
2017年, Gunasekar 等[45]合成出Ir氮杂环卡宾配合物的共价三嗪骨架材料Ir-NHC-CTF(图8). 作者为了验证合成的催化材料的性能, 将其用于CO2氢化制备甲酸催化反应中. 结果表明, Ir-NHC-CTF对CO2加氢生成甲酸反应具有良好的活性, TOF与TON值分别可以达到16 000 h-1和24 300, 这是当时报道出来的催化剂在多相催化体系的最高值. 也足以证明了该多相催化剂在催化反应中具有极其巨大的潜力.
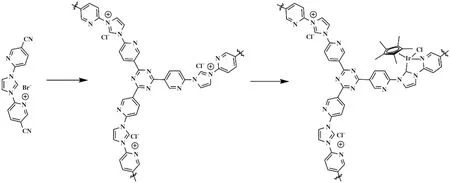
图8 Ir-NHC-CTF的合成[45]Fig.8 Synthesis of the Ir-NHC-CTF[45]
同样利用NHC配体富电子的特殊性质, Li及其同事[46]将其引入到共价三嗪骨架材料中用于催化CO2硅氢化制备甲酰胺的环加成反应, 作者证明了含内嵌NHC的三嗪连接聚合物在CO2吸附方面展现出优秀的性能, 更重要的是通过形成两亲性加合物NHC-CO2, CO2被原位激活后由此产生的NHC-CO2-Triazine聚合物具有较高的活性和稳定性.
为了实现催化位点的均匀分布与催化剂高负载量的目标, 2020年, Li及其同事[47]通过溶剂热条件直接聚合的方式合成出NHC-AuCl-COF催化剂材料, 并且可继续通过阴离子交换策略制备出NHCAuSbF6-COF. 这两种多相催化剂用于催化末端炔与CO2的羧化反应和炔的水合反应. NHC-AuCl-COF使用DMF或DMSO作溶剂时, 温和条件下催化CO2羧化合成羧酸均能达到96%的产物收率. 同时, 作者用不同底物探究该反应发现大多数都能实现良好的产率(95% ~ 99%). 而相比之下, NHC-AuSbF6-COF催化剂对炔烃水合反应的作用尤为突出. 在优化的反应条件下, 催化苯乙炔和水反应生成苯乙酮的产率高达99%, 此外, 对其它一系列末端芳香炔的底物都表现出优异的催化活性.
2019年, Zhang等[48]通过原位生成策略将咪唑基连接剂掺入到MOF中, 利用咪唑脱质子形成自由的NHC复合物与ZrII-MOF结合构筑UiO-68-NHC催化剂(图9), 能在极其温和的反应条件下(0.1 MPa,室温)下, 用CO2将硅烷定量转化为甲醇, 其产物通过水解继续反应最终全部转化为甲醇. 而作者采用DFT计算也证实了NHC位点的作用有利于吸附硅烷前体, 同时硅基甲氧基产物分子与MOF单元的苯环之间存在π-π相互作用而更倾向于扩散到MOF孔道. 并且与均相的NHC基催化剂比较, UiO-68-NHC具有更优异的活性与选择性.
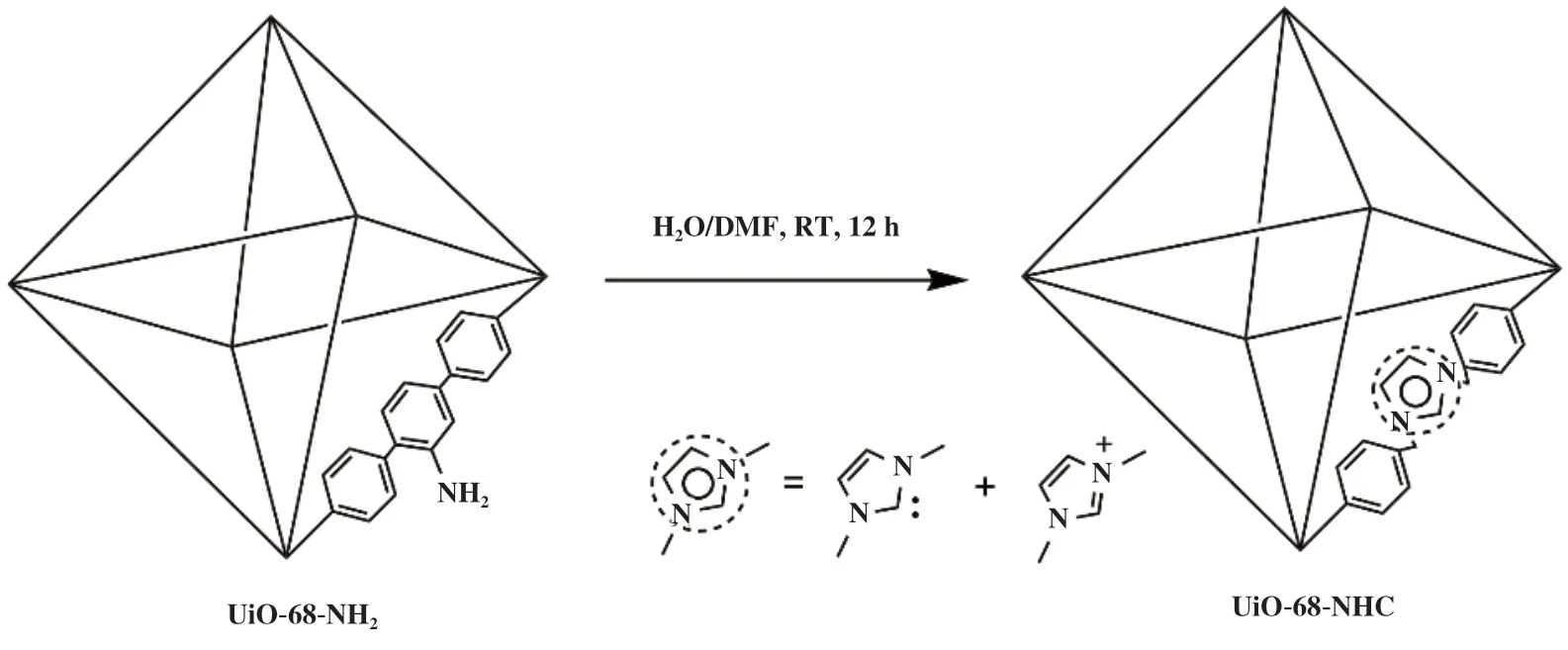
图9 UiO-68-NHC催化CO2硅氢化反应[48]Fig.9 Hydrosilylation of CO2 catalyzed by UiO-68-NHC[48]
3 C-C偶联反应
碳-碳键(C-C)的形成是目前最重要且常见的有机反应之一, 在材料学、 催化以及天然产物等领域得到了广泛应用. 由于其在工业和合成化学中的意义重大, 已有研究学者对C-C交叉偶联技术进行详细的论述[49]. 而金属Pd催化的C-C偶联反应凭借着高活性和高选择性而成为首选, 但同时, 均相过程所带来的催化剂从产品中分离回收困难等问题也会出现[50]. 因此, 在MOFs和COFs载体上固定化Pd配合物能够表现出良好的稳定性和重复利用性. 例如, 2011年, Ding 等[51]首次利用COFs材料催化Suzuki反应, 证明了COFs材料在催化领域有着无法预测的作用, 也正是他们对COFs多孔材料作出的突出性贡献, 使得更多的科学家们对未来MOFs和COFs材料在多相催化中的研究也有了重新的定义.
在开发稳定基体负载Pd多相催化剂的同时, 经济高效的载体与强金属-载体相互作用的制备策略也固然重要. 为此, 在类似MOFs或COFs的多孔聚合物中引入氮杂环卡宾(NHC)基团对解决金属催化中心分布不均匀和金属-载体间的弱相互作用所导致的金属聚集和泄露问题具有重要意义[52]. NHCs作为一种高效的功能化配体具有独特的自身优势,例如强σ-donor能力、 在空气和水中稳定性优异以及毒性小等特性. 同时, NHCs配体能与金属形成较强的M-C键并且可根据调节不同的金属位点显著提高催化活性而受到众多化学家的青睐[53-54].
2018年, Dong课题组[55]报道了一种新型的多相催化剂(UiO-67-Pd-NHDC), 他们是利用配体前修饰的方法先合成出含金属Pd的氮杂环双卡宾二羧酸配体(Pd-NHDC-H2L), 接着在溶剂热条件下与尺寸匹配的ZrCl4和双苯基-4,4’-二羧酸结合而成.UiO-67-Pd-NHDC用于催化Heck偶联反应具有较好的催化活性(图10), 使用碘苯与丙烯酸乙酯作为底物, 用NEt3作为碱源, 在1 h内可以得到99%的产物收率. 此外, 在优化的反应条件下, 作者探索不同底物之间的催化性能发现由于该催化剂在内外表面是共同作用的, 从而导致在卤素取代的芳基化合物中,芳基碘化物具有更好的反应性.

图10 UiO-67-Pd-NHDC催化Heck偶联反应[55]Fig.10 Heck coupling reaction catalyzed by UiO-67-Pd-NHDC[55]
为了避免催化过程中反应温度高或反应时间长所带来的困扰, Yang等[56]率先设计合成出功能化亚胺连接的COF材料(COF-NHC), 并利用NHC配体与金属的强结合能力与Pd(OAc)2反应制备出NHC功能化的Pd(II)配位的COF催化剂(Pd@COF-NHC).在催化卤代苯与4-甲基苯基硼酸为原料的Suzuki偶联反应时发现, 在室温条件下以水作为介质, 1 h的时间该反应不但能够发生而且所得的产率高达99%, 这个惊人的结果是在此之前从未有过报道的.他们继续用水为介质催化不同反应底物的Suzuki反应, 都表现出优异的催化性能(95%~99%). 值得注意的是, Pd@COF-NHC催化8次循环后催化活性并无明显损失, 这足以说明其具有超强的稳定性能.
除了配体预修饰方法合成含Pd-NHC配合物以外, 后合成修饰法也有学者报道过. 2017 年,Bahadori及其同事[57]采用合成后改性法制备出氮杂环双卡宾固定的MOF材料, 并实现了与Pd原位配位形成MIL-NHC-Pd多相催化剂. 为了研究其催化活性, 作者以苯基硼酸和芳基卤化物为合成原料的Suzuki反应对催化剂进行评价. 在温和的反应条件下, MIL-NHC-Pd具有较好的催化效率; 同时,在反应之后对催化剂进行回收再利用可连续运行15次.
2020年, Nezhad团队[58]使用类似的合成方法通过MIL-101氯甲基化反应、 接着与1-甲基咪唑反应构建氮杂环卡宾片段并负载Pd(OAc)2制备Pd-NHC-MIL-101(Cr)催化剂(图11). 通过用4-甲氧基溴苯和苯硼酸做底物筛选出最佳反应条件, 结果发现用H2O作溶剂, K2CO3作碱, 85 ℃时很短的时间内催化Suzuki反应能达到90%产率. 同时由于该催化剂催化位点存在于微孔内外, 对合成联芳基分子也产生了一定的作用. 由于NHC与Pd存在强配位作用,使催化剂体系具有强稳定性, 运行8次后仍然保持活性. 随后, 作者还提出了Pd-NHC-MIL-101(Cr)催化剂体系进行Suzuki反应主要分三步: 第一步是芳基卤化物(Ar-X)与Pd(0)配合物氧化加成形成Ar-Pd(II)-X复合物; 其次, Ar-Pd(II)-X再与金属化过程活化后的芳基硼酸[Ar-B(OH)3]反应连接苯环; 最后, 通过还原消除反应得到偶联产物. 此外, 再生的Pd(0)物种继续催化下一个循环.

图11 Pd-NHC-MIL-101(Cr)的合成[58]Fig.11 Synthesis of the Pd-NHC-MIL-101(Cr) [58]
最 近, Niknam 等[59]制 备Pd-NHC-MIL-101(Cr)催化剂用于其它类型C-C偶联反应, 在优化的反应条件下, 对Heck反应具有良好的底物适用性, 可观的偶联产物收率得以实现. 与此同时, 催化剂也在无铜Sonogashira偶联反应中表现出较高的活性与催化效率, 能保证获得多种炔烃衍生物产物.
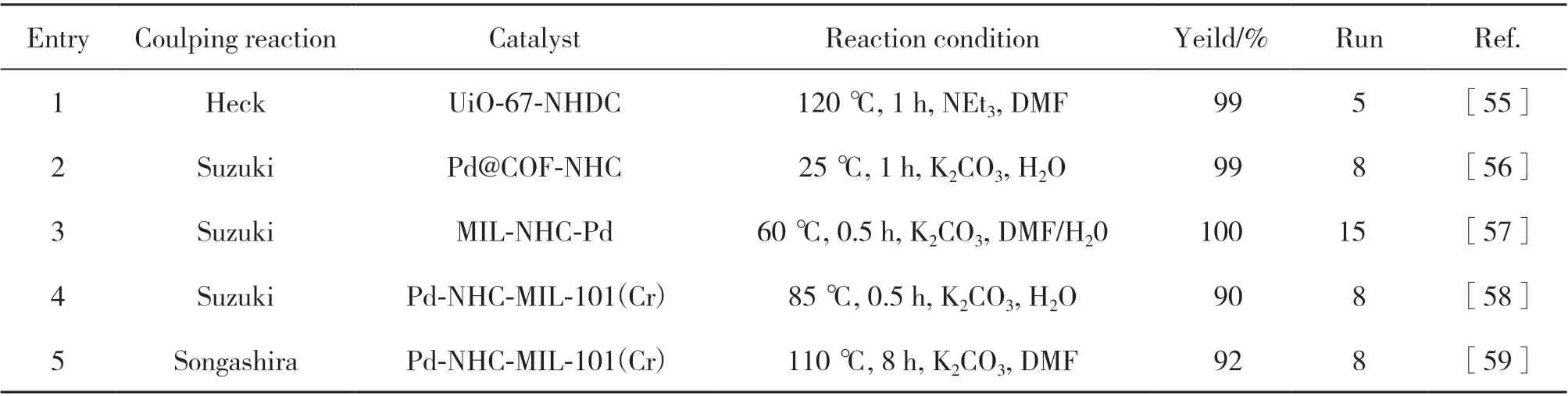
表2 含IL的MOFs和COFs催化体系催化C-C偶联反应的总结Table 2 Summary of C-C coupling reations catalyzed by IL-containing MOFs and COFs catalytic systems
4 羰基化反应
羰基化反应(carbonylation)是指在催化剂作用下, 将羰基或其它基团引入到不饱和烃、 醇、 酚、 环氧化合物等各类反应底物分子中而形成含氧化合物的一类重要反应, 该反应最初是由德国化学家Otto Roelen于1938年研究费-托合成时首次发现的[60].经过几十年来的发展, 羰基化反应所催化出的产物遍及众多化学品以及聚合物材料. 更重要的是, 由于其具有原子经济性独特优势, 在C1资源的高值化利用方面逐步受到众多化学工作者的青睐[61-62]. 然而, 羰基化反应所存在的贵金属流失与分离回收等缺陷也是不可避免的. 近年来, 为了能够解决上述问题并能够有效提高催化效率, 研究人员将离子液体或离子液体固载化的框架材料引入到羰基化反应中[63]. 而截止到目前, 只有少量文章报道了含离子液体片段的框架材料用于甲醇羰基化反应和环氧化合物羰基化反应中.
2017年, Yoon课题组[64]先后合成了含咪唑离子液体的氰基单体, 并通过离子热聚合方法在ZnCl2作为助催化剂的情况下, 将含三嗪结构的聚合物材料作为载体, 与Co(CO)-4离子形成配合物得到第一种离子液体基多相钴催化剂[imidazolium-CTF][Co(CO)4](图12). 该催化剂用于环氧丙烷直接开环羰基化合成3-羟基丁酸甲酯(MHB)反应中, 在优化的反应条件下, MHB的选择性高达86%. 此外,[imidazolium-CTF][Co(CO)4]非均相催化剂在5次循环利用后仍能保持85%活性并能进行有效的羰基化反应.

图12 [imidazolium-CTF][Co(CO)4]的合成[64]Fig.12 Synthesis of the [imidazolium-CTF][Co(CO)4] [64]
随后, 他们在研究中发现虽然咪唑钴酸盐多相催化剂在催化环氧化合物羰基化合成β-羟基酯时能够表现出优异的活性与选择性, 但同样不可避免的是催化剂载体中钴酸盐的浸出也很大程度地限制了其重回收与利用. 为了克服这些缺点,Rajendiran 等[65]改用双咪唑基共价三嗪骨架作为环氧丙烷羰基化反应的载体, 并继续使用K2Co(CO)4作为金属来源与载体反应制备钴基多相催化剂[bisimidazolium-CTF][Co(CO)4]xCl(2-x), 希望通过咪唑部分多个阴离子的稳定作用从而达到更优的选择性和可回收性. 而结果不出所料, 该催化系统在催化环氧丙烷羰基化反应中, 其性能有显著提高, MHB的产率可高达93%(表3). 此外, 由于阴离子的稳定作用, 该催化剂循环6次后仍保持95%的活性.
证实了Co金属固载离子液体基框架材料在羰基化反应的高效利用后. 2018年, Yoon课题组[66]又成功地合成出一个带电荷的1,3-双(吡啶基)咪唑基共价三嗪骨架基于铑的分子催化剂(Rh-bpim-CTF),在原料气CO∶N2为9∶1的压力下在反应器中进行甲醇的羰基化反应. 该催化剂表现出优异的甲醇转化率(93%), TOF值高达2100 h-1. 令人意外的是, 当提高比例时还会表现出更好的催化性能, 对连续的甲醇羰基化具有极好的价值.
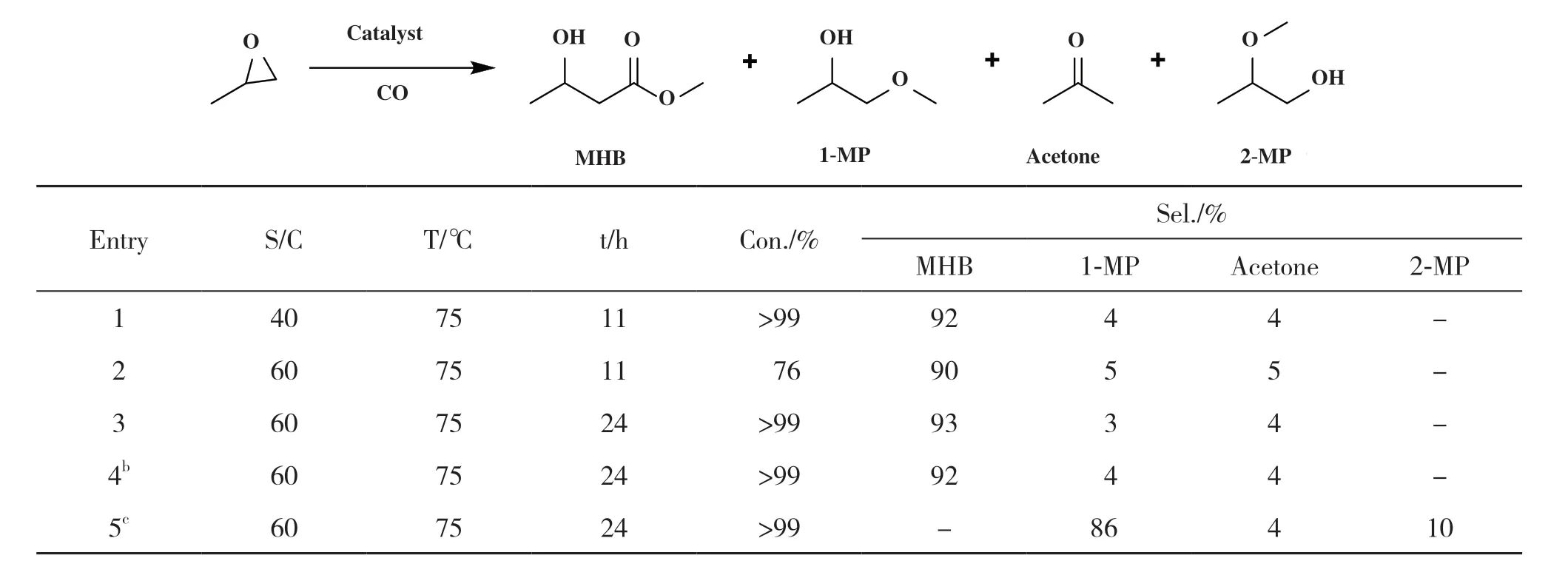
表3[ bis-imidazolium-CTF][Co(CO)4]xCl(2-x)的催化性能a[65]Table 3 Catalytic ability of using[ bis-imidazolium-CTF][Co(CO)4]xCl(2-x)a[65]
5 其它有机转化反应
近些年来, 除了上述介绍到的CO2利用反应、Pd催化C-C偶联反应以及羰基化反应以外, 用离子液体功能化的框架材料作为载体负载金属用于各类催化反应都已经有所报道. 为了能够证明离子液体功能化的框架杂化材料在催化领域的广泛性, 我们将各类催化反应逐一列举出来.
2013 年, Luo等[67]通 过 在MIL-101 纳 米 笼 中容纳酸性季铵盐离子液体制备出TEDA-BAIL/MIL-101多相催化剂, 在苯甲醛和乙二醇为原料的缩醛反应中进行了催化性能评价, 优异的催化活性(88%转化率与100%选择性)得益于离子液体催化剂与MIL-101纳米笼内良好的微环境相结合. 此外, 催化剂能够循环使用6次而没有明显的活性降低. 随后, Luo 等[68]又探究了HKUST-1 型MOF材料分散到氨基功能化的碱性离子液体 1-甲基-3-(2-氨基乙基)咪唑在不同溶剂(H2O、 EtOH、 DMF)中的作用和催化行为. 整个过程可通过简单的室温搅拌1 d、 溶剂洗涤和真空烘干步骤完成. 由此合成出的ABIL@HKUST-1多相催化剂用于催化苯甲醛和丙二腈的Knoevenagel缩合反应. 结果发现, 以EtOH为溶剂制备出来的催化剂具有最佳的催化性能(100%转化率).
最近, Gao课题组[69]使用含苯胺单体与两个等摩尔比的线性双醛单体为原料制备亚胺连接的COF,再结合溴代烷烃发生季胺化反应与加碱去质子化过程最终实现多相催化剂(NHC-Py-COF)的生成, 催化甘油与碳酸二烷基酯交换反应具有良好的催化性能, 反应温度70 ℃, 8 h条件下, 催化甘油与碳酸二甲酯或者碳酸二乙酯都表现出较高的转化率(99%或96%)与产物收率(96%). 此外, 催化剂可重复利用性也十分可观, 5次循环后不会造成明显的损失.
2021 年, Zhang团队[70]通过将乙烯基修饰的COF与4-乙烯基苄基氯(VBC)聚合, 然后再与叔胺发生季胺化反应设计出一种新型的杂化材料PILCOF-X. 为了证明催化活性位点更好地容纳反应物,对材料进行的分析结果表明, 在适当的总醛边单元值X=0.33时, 合成的PIL-COF-0.33杂化材料保留了原有的结晶度和孔隙度. 随后对其杂化材料在山梨醇脱水转化异山梨醇催化反应进行了评价, 在优化的反应条件下, PIL-COF-0.33表现出优异的活性和选择性. 并在同样的条件下进行催化剂回收实验,异山梨醇的产率在连续10次运行后仍相对稳定在77%以上.
目前为止, 合成小尺寸、 分散均匀和可重复利用的金属纳米颗粒(NPs)仍然是具有挑战性的任务,而富N环境的多孔三嗪聚合物与离子液体原位生成的NHCs可以起到稳定NPs作用, 并保持良好的催化性能. 2021年, He等[71]为了克服串联反应耗时长与成本高所带来的的困扰, 首次开发了具有多个活性位点功能化的催化剂体系. 他们利用咪唑修饰的阳离子共价三嗪框架独特的Lewis酸碱作用与AuNPs进行串联反应, 得到的多功能催化剂Au@ICTF于温和的条件下, 在催化串联脱醛-Knoevenagel缩合-还原反应中表现出良好的催化活性与选择性(图13).
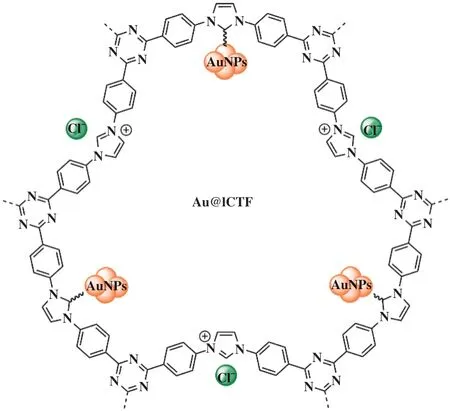
图13 Au@ICTF催化的串联反应[71]Fig.13 Tandem reaction catalyzed by Au@ICTF[71]
2021 年, Mao等[72]利用点击化学原理, 通过Debus-Radziszewski咪唑合成反应制备出具有高比面积的多种咪唑基多孔三嗪聚合物材料(IPTP), 并通过离子交换策略将PdCl2-掺和到咪唑基聚合物框架中并还原得到Pd/IPTP催化剂. 结合表征手段发现, 框架中丰富的咪唑基团和三嗪单元有助于促进Pd纳米粒子的稳定性, 同时三嗪结构作为双功能位点也可在催化反应中起到加速催化作用, 显著提高催化活性. 对制备的Pd/IPTP在苯甲醇脱氢制苯甲醛时的催化性能进行评价, 结果表明, 反应条件最佳时表现出优异的催化性能(转化率为98%, 选择性为99%).
在各种金属催化的有机反应中, Cu纳米材料作为一种经济有效的金属, 也通常被认为是最有效的催化剂金属之一. 最近, Firouzeh团队[73]使用咪唑离子液体作为含氮的前驱体与三聚氰氯作用, 最后在离子热过程中成功构建出共价三嗪介孔固体载体材料CTF, 并使用Cu(NO3)2和N2H4在载体表面实现了Cu纳米颗粒固载化, 所制得的多相催化剂Cu@CTF对硝基芳烃加氢反应具有良好的催化性能. 作者使用硝基苯作为反应底物, NaBH4为还原剂, 在温和的反应条件下(水为溶剂, 60 ℃, 25 min)催化硝基苯加氢制苯胺反应高达100%转化率. 此外, 该催化剂具有高结构稳定性, 可回收和重复使用7次且活性没发生明显降低现象.
6 结论与展望
我们系统总结了近年来在离子液体功能化金属有机框架MOFs和共价有机框架COFs杂化材料作为催化剂或作为金属纳米颗粒或者氮杂环卡宾金属配合物载体用于CO2和环氧化合物的环加成、 CO2还原功能化应用、 C-C偶联和羰基化等各类催化反应, 这些现有工作极大地丰富了离子液体修饰的多孔有机聚合物材料在催化方面的应用. 同时部分解决了目前上述反应存在的分离回收困难、 反应条件苛刻和催化活性及选择性低等难题. 但是, 该领域仍有许多亟需解决的问题, 如: (1) 多数催化剂的综合性能不够理想, 制备步骤繁琐以及结构稳定性差等缺陷, 严重限制了其大规模应用; (2) 对离子液体功能化金属有机框架和共价有机框架杂化材料催化的一些特定反应的催化机理需要更深入地研究,阐明MOFs、 COFs和离子液体片段的有效功能; (3)设计和开发新型结构离子液体功能化金属有机框架MOFs和共价有机框架COFs复合催化材料, 有效拓展在其它类型催化反应中的应用也是未来发展的重要方向之一.
