利用CRIS PR/Cas9 技术创制水稻中花11 号dwarf1 突变体
2023-01-13冯彦钊朱庆锋
冯彦钊,朱庆锋,薛 皦,陈 沛,于 洋
(广东省农业科学院农业生物基因研究中心/广东省农作物种质资源保存与利用重点实验室,广东 广州 510640)
【研究意义】随着人口的迅速增长与全球气候变化,植物的生长、抗性和品质等受到严峻挑战。分子育种是改良目标性状、获得更加优质作物的强力手段。基因编辑是一种利用核酸酶对目标基因进行碱基定点删除、插入、替换或修饰的技术,利用该技术对特定的基因进行改造可以创制新的种质[1-2]。水稻中花11 号是由中国农业科学院作物科学研究所育成的优质水稻品种,不仅具有良好的产量和抗逆性,同时也是植物分子生物学基础研究领域最具代表性、使用最广泛的模式品种之一。DWARF1(D1)是编码水稻三聚体G 蛋白α 亚基的基因,调控水稻多种生命活动过程[2-3]。然而,在中花11 号中仍缺少对应的d1缺失突变体,给G 蛋白相关通路的遗传学研究带来诸多不便。
【前人研究进展】早期的基因编辑技术利用大范围核酸内切酶诱发双链断裂,从而产生随机的碱基插入/缺失或同源重组。然而这种内切酶切割效率较低,且需要长片段的识别序列[4-5]。随后出现的锌指核酸内切酶(Zinc Finger Nucleases,ZFN)利用氨基酸模块与DNA 三连续碱基一一对应的关系进行靶向的DNA 切割,基因编辑效率大大提高[6-8]。不久,人们从侵染植物的黄单孢菌中获得启发,利用类转录激活效应因子核酸酶(Transcription Activator-like Effector Nucleases,TALEN)作为内切酶进行碱基编辑[9-10]。与之前的ZFN 相比,TALEN 中一个氨基酸模块只对应一个碱基对,使设计更加简易[11]。尽管如此,ZFN 和TALEN 均需要针对不同的靶点序列设计合成相应的蛋白,较耗时耗精力。CRISPR/Cas9 则是新一代基因编辑技术,最突出的优点是不依赖于蛋白质识别靶点DNA 序列,而是通过引导RNA(single guide RNA,sgRNA)与靶点DNA 互补配对,Cas9 识别前间隔序列邻近基序(Protospacer Adjacent Motif,PAM)并行使核酸酶活性对靶基因进行切割[12-14]。至今,基于Cas9 的技术已经被开发出多种形式,可以实现基因激活、荧光定位、单碱基编辑、定向编辑等[15-18]。Cas9 的同家族蛋白Cas12 和Cas13 也相继被开发用作编辑DNA 和RNA[19-20]。由于该技术的简易性,CRISPR/Cas9 已成为应用最广的基因编辑技术,在基础研究和应用领域创制了大量宝贵材料[21-25],在改良农艺性状上有广阔的应用前景[26-27]。
异源三聚体G 蛋白是定位于动植物细胞膜表面的重要信号转导因子,作用于受体下游。它由α、β 和γ 共3 个亚基组成,在植物中参与激素信号转导、离子通道调控、免疫应答、细胞分裂等关键生理进程。与动物不同的是,植物基因组G 蛋白α 亚基拷贝数非常少,水稻只有一个编码G 蛋白α 亚基的基因D1。D1基因有许多重要功能,与Gβ 和Gγ 组成复合体,调控水稻对氮素的利用和种子大小[34-38]。此外,D1在稻瘟病的抗性中,与OsRac1共同调控OsMAPK6的蛋白水平,影响下游防御基因的表达[39]。在淹水胁迫环境中,D1在乙烯诱导缺氧薄壁细胞程序性死亡的过程中是必需的[40-42]。同时,D1也是油菜素内酯和赤霉素中信号转导通路中的一员[43]。运用突变体对D1进行研究已有相当长的历史,但其突变体遗传背景局限于日本晴、Taichung65(T65)和Kinmaze 中[2,44]。日本晴背景的d1突变体DK22 第5 个外显子G1721T 的点突变产生终止密码,导致较短和较圆的籽粒以及对赤霉素和油菜素内酯的敏感性降低[45]。同为日本晴背景的HO541 有833 bp 缺失,出现短且深绿的叶片,花序变得更紧凑[2]。Kinmaze 背景下的CM1361-1 在D1的第5 个外显子中发生19 bp 插入,导致节间变短[3,45]。T65 背景d1突变体中的D1基因最后一个外显子发生2 个碱基缺失,对稻瘟病更易感[3]。中花11 号作为我国水稻基础研究常用的品种之一,遗传转化体系非常成熟,许多基因在此背景中都有相应的突变体,极大促进了水稻基因功能研究。
【本研究切入点】构建有效的中花11 号背景下的d1突变植株,将为水稻基因功能研究提供便利。利用CRISPR/Cas9 技术对中花11 号野生型植株D1基因位点进行编辑,可以在中花11号背景中产生新的D1等位突变,丰富现有的d1突变体种质。【拟解决的关键问题】本研究利用CRISPR/Cas9 技术编辑水稻中花11 号的D1基因,筛选及鉴定无抗、有效移码突变的d1突变植株,并分析d1突变体的表型,以期为进一步研究D1的未知功能提供宝贵材料。
1 材料与方法
1.1 试验材料
本研究所用水稻材料为中花11 号,为广东省农业科学院农业生物基因研究中心分子育种技术研究室保存;所用大肠杆菌感受态为Trans1-T1 Phage Resistant Chemically Competent Cell,购自北京全式金生物技术有限公司;所用农杆菌感受态为EHA105,购自上海唯地生物技术有限公司。试验地点为广东省农业科学院农业生物基因研究中心,试验时间为2021 年3 月至2022 年9 月。
1.2 靶向D1 的CRISPR/Cas9 载体构建与遗传转化
利用He 等[46]报道的pEntry A 与pRHCas9载体进行构建。基因编辑靶点用CRISPR-P 2.0 进行筛选(http://crispr.hzau.edu.cn/CRISPR2/),选择的靶点为5'GCTTTGATGAGGCAGAACTT3',靶点在基因组中的位置为625~644(图1B),合成引物(表1)。以pEntry A 为模板,用ENTRY-F/D1-gRNA-41R、D1-gRNA-41F/ENTRY-R 扩增成两个片段。将这两个片段以摩尔比1∶1 混合,再以ENTRY-F/ENTRY-R 引物进行第二轮扩增。反应程序为98 ℃ 3 min;98 ℃10 s、60 ℃ 30 s、68 ℃ 30 s,35 个循环;68 ℃终延伸5 min。第二轮扩增的产物经琼脂糖凝胶回收后,按照He 等[46]方法连接至pRHCas9 中。构建好的载体转入大肠杆菌感受态,提取质粒,测序比对后再转入农杆菌感受态,参照崔莹等[47]方法侵染水稻胚愈伤组织,获得转化植株。
1.3 DNA 提取及PCR 检测
DNA 提取参考王齐红等[48]方法进行。基因型检测的引物为D1-CRISPR-926F71/D1-CRISPR-926R69,利用KOD FX 聚合酶扩增,反应程序为98 ℃ 3 min;98 ℃ 10 s、68 ℃ 60 s,35 个循环;68 ℃终延伸5 min。潮霉素、载体骨架和ACTIN基因检测引物分别为HPT-F/HPT-R、VEC39-315F74/VEC39-315R75、ACTIN-F/ACTIN-R,序列如表1 所示。反应程序为 98 ℃ 3 min;98 ℃ 10 s,62 ℃ 30 s,68 ℃ 60 s,35 个循环;68 ℃终延伸5 min。
1.4 RNA 提取与qRT-PCR 检测
按照Simms 等[49]方法进行RNA 提取,利用PrimeScript™ RT reagent Kit with gDNA Eraser(Perfect Real Time)(RR047)进行反转录,以TB Green®Premix Ex Taq™ II(Tli RNaseH Plus)(RR820)进行荧光定量PCR,D1的qRT-PCR引物为D1-qF/D1-qR,内参基因引物为UBQ5-qF/UBQ5-qR,序列如表1 所示。反应程序为95 ℃ 30 s;95 ℃ 10 s、60 ℃ 30 s,40 个循环。相对表达量用2-〔d1(CtD1-CtUBQ5)-WT(CtD1-CtUBQ5)〕计算。显著性分析采用Student’st-test。

表1 供试引物及其序列Table 1 Primers and their sequences used in this study
1.5 表型测量与统计
株高用自地面到主穗顶长度的平均值表示,n=10。穗长用穗颈节到穗顶的长度平均值表示,n=15。粒长、粒宽分别为饱满种子的长度和宽度平均值n=12。千粒重用1000 粒饱满种子的质量平均值换算而成,n=20。显著性分析采用Student’st-test。
2 结果与分析
2.1 水稻中花11 号d1 突变体的创制
为创制中花11 号背景下的d1突变体,我们采用CRISPR/Cas9 基因编辑技术对D1基因进行突变。首先选取位于第二个外显子上的一段序列为靶点序列(见1.2)作为靶点,此位置与已报道的D1突变位点均不相同。将靶点序列正向引物与下游gRNA 的5'端序列融合,sgRNA 反向引物与上游U6 启动子的3'端融合,再与pEntry A载体上的引物进行融合PCR,得到U6 启动子-靶点-gRNA 表达盒。将扩增产物与pRHCas9 载体用PstI 和SpeI 双酶切,再进行连接得到D1-Cas9 载体(图1A)。将D1-Cas9 载体导入农杆菌EHA105 中,侵染ZH11 愈伤组织,随后通过共培养、筛选、分化等步骤得到d1突变体幼苗。
2.2 突变体基因型分析及表达水平检测
为分析Cas9 是否对D1基因位点进行编辑,我们对d1突变体T1代植株进行DNA 提取,以其为模板,在编辑位点上下游400~500 bp 处设计引物进行PCR 扩增(图1B),对产物进行Sanger测序,将测序结果与WT 进行比对。发现突变体的D1基因位点发生编辑,3 个株系均为纯合移码突变,其中d1-#1为-17+7 bp,d1-#2为+2 bp,d1-#3为+1 bp(图2A)。为了解突变株中D1的表达水平,我们对T2代纯合突变植株的幼苗进行RNA 提取并采用荧光定量PCR 检测D1的mRNA水平。结果显示,3 个突变株系D1的mRNA 水平均显著下调,下调幅度高达80%~90%(图2B)。这说明d1突变株在mRNA 和蛋白两个水平上抑制D1的功能,成功获得有效的d1突变植株。
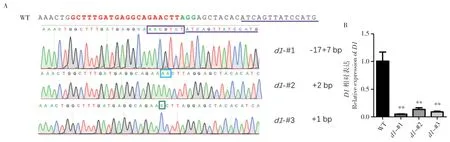
图2 d1 突变株的基因型鉴定及D1 表达量检测Fig.2 Genotyping of d1 mutants and detection of expression level of D1
2.3 无潮霉素抗性突变体筛选
转基因标记是作物向农业生产应用的障碍,易引发公众对转基因安全性的忧虑。同时,转基因产生抗性标记的存在可能为以后基础研究所需的杂交带来不便。如两亲本抗性相同,难以用抗性准确筛选杂交成功的后代植株。因此,筛选无潮霉素抗性的d1突变株具有重要意义。农杆菌介导转基因通常以单拷贝形式随机插入到基因组中,通过 自交产生T1代时,抗性基因可通过同源重组或自由组合分离,得到无抗性的突变体。利用该原理,我们对T2代植株叶片的DNA进行提取,并且通过PCR 扩增HPT外源基因片段,同时扩增了ACTIN启动子片段作为内源基因的参照。结果显示,每个株系随机选取的若干植株中,均分离出不含外源基因的突变株(图3),为后续研究与应用奠定基础。
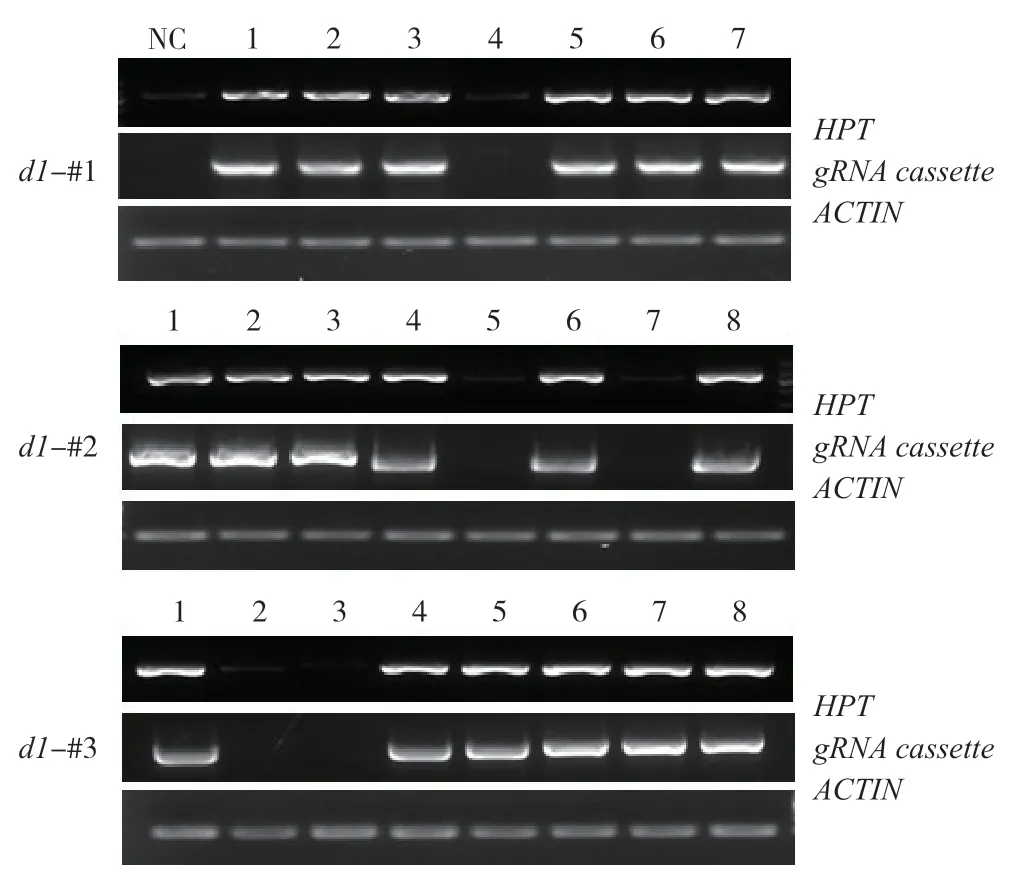
图3 d1 各突变株系不同植株DNA 中转基因标记的PCR 检测Fig.3 Detection of transgenes in genomic DNA of d1 mutants by PCR
2.4 d1 突变体表型分析
以往研究表明,d1突变体的株型、穗型、籽粒形态与大小等与野生型有明显差异。为全面了解中花11 号背景下d1突变体的表型,我们对植株形态、穗形和种子大小进行观察(图4)与统计(图5)。统计结果显示,3 个d1突变株系均显著矮化,株高仅有野生型的约50 %。此外,穗长只占野生型的约60%。d1突变体的种子大小、形态同样发生明显变化,其中粒长变得更短,野生型平均粒长为0.74(±0.05)cm,而3 个d1突变株系分别为0.55(±0.06)、0.62(±0.03)、0.55(±0.04)cm。然而,粒宽在d1突变体中无明显变化,因此,d1突变体种子变得更圆,长宽比为1.29(±0.16)、1.46(±0.12)、1.34(±0.15),中花11 号的种子长宽比则为1.85(±0.10)。同时,d1植株千粒重显著降低,约为野生型的60 %(图5F),可见中花11 号背景下d1突变体产生严重的生长缺陷。

图4 d1 突变株的形态特征Fig.4 Morphological characteristics of d1 mutants
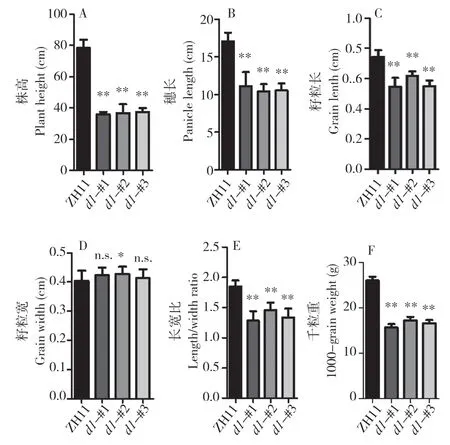
图5 d1 突变株的表型统计Fig.5 Phenotype statistics of d1 mutants
3 讨论
D1是植物生长发育过程中一个重要的调控基因,日本在20 世纪初期已有该基因的水稻突变体。D1基因功能具有多效性,近年陆续出现其他背景下的D1等位突变体,如T65、Shiokari、Kinmaze。这些突变体突变形式各异,包括点突变、碱基插入/缺失导致蛋白翻译提前终止、结构域失活等,也有剪接位点改变和反义转录本导致的RNA 水平降低[44]。此外,科学家还发现了天然的表观遗传d1突变体,其DNA 序列并没有发生变化,然而矮化的d1表观突变体转录起始位点附近DNA 发生甲基化,同时,在D1基因座内,组蛋白H3K9 乙酰化水平降低而H3K9 二甲基化水平升高,表明d1表观突变体D1的表达被沉默[50]。在双子叶模式植物拟南芥中,D1同源基因的缺失突变体已有4 个株系被报道,它们属于Ws和Col背景[51-52]。其他植物种类如大豆、烟草、西红柿等的D1同源基因功能研究并不深入,多数仅停留在基因的克隆、鉴定阶段,缺乏相应的功能缺失突变体[53]。本研究采用CRISPR/Cas9 介导的基因编辑技术对中花11 号的D1基因目标位置进行编辑,鉴定出3 个等位突变株系,丰富了现有的d1突变体类型。
现有研究表明,水稻d1突变体会导致植株矮小、直立,叶长变短且深绿、谷粒变短变圆、节间变短、对GA 和BR 敏感性降低、免疫应答改变等多效表型[44]。我们创制的中花11 号背景下的d1突变体也出现植株矮小、穗小、籽粒变短、籽粒变薄等表型,与前人报道的其他背景下的d1突变体表型相吻合,表明D1在不同水稻品种中的功能高度保守,也证明本研究所得到的材料可有效应用于相关领域研究。
CRISP R/Cas9 基因编辑产生的移码插入或缺失突变通常引起翻译提前终止,导致无义RNA降解(Nonsense mRNA Decay,NMD)[54]。本研究所创制的d1突变体材料中,D1基因的mRNA水平下调约80%,我们分析d1突变体的编辑情况发现,d1-#1和d1-#2突变体均使D1 蛋白在第94 位氨基酸位置处提前终止,而d1-#3突变体会使D1 蛋白翻译在第35 氨基酸处提前终止,这些因素很可能触发mRNA 质量监控机制,导致D1mRNA 经历NMD 而下调,这些结果与我们之前在其他基因的CRISPR 敲除植株上观察到的相似[55]。然而,CRISPR/Cas9 导致NMD 并不是绝对的,与sgRNA 到下游外显子或终止子的距离远近有关[56]。可见,CRISPR/Cas9 介导的基因编辑有可能从mRNA 水平和蛋白水平两方面导致基因功能失活。
4 结论
本研究利用CRISPR/Cas9 技术在中花11 号背景中创制出3 个移码突变的d1突变株系,qRT-PCR 结果显示,D1mRNA 水平显著下调。中花11 号背景的d1敲除植株出现植株矮小、穗小、籽粒变小、变圆等表型,千粒重也显著减少,株高、穗长、粒长、长宽比和千粒重均下降40%~50%,而粒宽无明显变化,这些表型与其他背景中现有的d1突变体相似,说明D1基因功能已经失活。我们还从T2代植株筛选出不含转基因标记的植株。鉴于中花1 1 号基因组已被研究得非常透彻,被广泛用于基因功能研究,现有的突变体库也以它为背景创制[22,23],拥有中花11 背景的d1突变株对创制D1与其他基因的多重突变株更加便利。因此,本研究结果可以为后续的水稻基础研究与育种应用提供安全、有效的遗传材料。
