表面结构与催化性能关系的模型催化实验
——以CuO/Cu2O纳米晶催化乙烯实验与量化计算为例
2023-01-10刘梓歌王炯涵李俍江国顺李红春吴强华张万群冯红艳吴红王钰熙
刘梓歌,王炯涵,李俍,江国顺,李红春,吴强华,张万群,冯红艳,吴红,王钰熙
化学国家级实验教学示范中心(中国科学技术大学),合肥 230026
催化剂表面结构与催化性能之间的关系一直是表面催化领域的前沿研究课题。该课题包含了催化反应动力学、热力学、表面化学、结构化学中重要的物化概念及实验方法,因此有必要引入到物理化学实验教学中。而在物化的传统实验教学项目中,因为实验条件的苛刻性或者催化剂表面结构复杂性等原因[1],这一重要课题鲜少被涉及,毫无疑问限制了学生对于催化剂结构及催化过程的进一步理解。近年来,科研工作者提出的基于形貌规整、大小均一、组成可控的纳米晶催化剂可以在实际催化反应条件下研究催化剂表面结构和催化性能的方法[2,3],使得将这一前沿课题纳入实验教学成为可能。
通过对催化剂活性进行测试,可获得催化剂宏观性能数据;通过量化计算研究催化剂微观性能,可获得原子层面上的催化剂表面结构信息和催化反应机理。实验与量化计算互相验证和补充,可建立起研究宏观催化性能与微观表面结构之间关系的通道。其中,量化计算已经是物理化学研究的必要手段,也是本科物理化学实验乃至物理化学教学中一个亟待补充完善的内容,我们在本实验中进行了量化计算与实验结果对比分析的尝试。
教学组选取了性质稳定、易于制备[4]的暴露(100)晶面和(111)晶面的立方体和八面体CuO/Cu2O纳米晶作为模型催化剂体系,同时基于绿色安全性的考虑,将科研工作中的一氧化碳实验体系[2,3]改为乙烯作为反应气的新实验体系,设计了结合催化剂活性测试和量化计算的创新综合实验。实验中测试了两种表面结构不同的催化剂对乙烯的催化氧化活性,通过Arrhenius公式计算表观活化能,判断两者表面活性位点的差异。随后通过搭建CuO/Cu2O晶面结构并判断活性位点,加强对催化剂微观结构的认识,由量化计算研究乙烯分子在催化剂表面的吸附情况,结合实验结果进行分析,进而从微观上理解表面结构对催化活性的影响。通过对纳米晶模型催化剂体系的实验与量化计算结果的综合讨论,掌握研究催化剂宏观催化性能-微观表面结构关系的方式方法。
1 实验部分
1.1 实验原理
1.1.1 活性测试实验
本实验采用流动法测定固相催化剂的活性,使用转化率评价催化剂的活性。转化率指反应物的转化量占引入反应器反应物总量的百分比。实验中,反应气为乙烯与空气的混合气体(乙烯/空气体积比为0.5%)。活性测试实验试剂、催化剂、实验仪器详细信息请见表1-表3。
在200-300 °C的温度范围,反应气能稳定存在。当反应气经过CuO/Cu2O纳米晶催化剂床层时,会发生以下反应:

实验通过氢火焰离子检测器(FID)检测乙烯含量。FID的工作原理是含碳有机物在氢火焰中燃烧时,产生化学电离,在电场作用下,正离子被收集到负极,产生电流,经过放大收集记录数据。对于在相同的反应条件下(同样的催化剂装量,反应物料的进料速度等),根据(1)式计算不同反应温度下的乙烯转化率χ。

其中[A]0为反应气在反应管入口处的乙烯浓度;[A]为出口的乙烯浓度。利用气相色谱分析,[A]0、[A]分别对应于色谱峰的峰面积S0和S。转化率又可表示为:

利用Arrhenius公式得到该催化反应的表观活化能Ea。具体计算过程如下:

即

其中,k为速率常数,Ea为表观活化能,R为气体常数,T为热力学温度,A为指前因子。

其中,γ为化学反应速率;A’为反应相关的常数。

其中,S为乙烯反应气体的流量,实验中在室温下控制流量为20 mL·min-1,即1200 mL·h-1;η为乙烯体积占比,为0.005;χ为乙烯催化氧化反应的转化率;ρ为乙烯的密度,标准状况下的乙烯密度为1.264 g·L-1,利用理想气体方程计算室温下(25 °C)的密度为1.158 g·L-1;m为催化剂质量,实验上称取0.05 g;Mr为乙烯的相对分子质量。
1.1.2 量化计算
量化计算已成为研究材料性质的重要手段之一,在材料催化机理的理解方面也发挥了越来越重要的作用。使用相关应用程序可实现密度泛函理论数值方法的计算,帮助我们从原子尺度上研究材料表面催化性质。SIESTA (Spanish Initiative for Electronic Simulations with Thousands of Atoms)是一个免费的计算软件,可用于分子和固体的电子结构计算和分子动力学模拟,基于其内在原理,通常可以在一般的工作站上模拟几百个原子的体系[5]。
基于自旋极化的密度泛函理论(DFT)方法[6],使用SIESTA软件包来完成所有的计算。在计算中,采用的是广义梯度近似方法(GGA)的Perdew-Burke-Ernzerhof (PBE)交换泛函[7]。为了分析乙烯分子在催化剂表面的吸附,建立2 × 2的CuO/Cu2O(100)和CuO/Cu2O(111)的超胞来分别模拟立方体和八面体CuO/Cu2O纳米晶晶面结构。2 × 2 × 1k点网格用于描述结构优化的布里渊区。网格的能量截断值设为150 Ry,结构优化的力收敛标准低于0.05 eV·Å-1(1 Å = 0.1 nm)。分子在催化剂表面上的吸附能Ead定义为:Ead=Etotal-Esurf-Emol,其中Etotal、Esurf和Emol分别代表吸附体系的总能量、干净催化剂表面的能量和吸附分子在气相中的能量。量化计算软硬件详细信息请见表4。
1.2 试剂和材料
表1 实验试剂

表2 催化剂信息
1.3 仪器和表征方法
1.3.1 催化剂活性测试

表3 实验仪器
1.3.2 催化剂表征仪器
JSM-6700F型场发射扫描电子显微镜(日本电子公司),大功率转靶X射线衍射仪(日本理学公司),ESCALAB 250型高性能电子能谱仪(Thermo-VG Scientific公司),JEM ARM-200F型高分辨透射电子显微镜(日本电子公司)。
1.3.3 量化计算软硬件

表4 量化计算软硬件
1.4 实验步骤和方法
1.4.1 催化剂表征结果学习
教学组提供的两组催化剂样品是通过参考文献[8]中方法合成得到,首先制备形貌规整的立方体与八面体Cu2O纳米晶,表面经过氧化重构后形成了一层CuO薄膜,最终得到具有核壳结构的立方体和八面体CuO/Cu2O纳米晶,分别记为CuO/c-Cu2O和CuO/o-Cu2O。该催化剂的相关表征结果会在讲义中给出,表征结果分析作为实验预习考核的一部分内容。
1.4.2 催化剂活性测试
在活性测试实验中,小组同学得到两组催化剂,分别是立方体纳米晶,记为CuO/c-Cu2O,和八面体纳米晶,记为CuO/o-Cu2O。催化剂的活性测试是在固定床式反应器上进行的,称取50 mg催化剂与50 mg SiO2均匀混合后转移进石英管中,反应气体为0.5%乙烯-空气混气,气体流速为20 mL·min-1,通过小组讨论,选择一系列温度梯度进行测试,每个温度下保温15 min后测量。
1.4.3 量化计算
1.4.3.1 建立结构模型
使用Materials Studio 8.0软件建立分子、催化剂表面和催化剂表面分子吸附的初始构型,包括建立乙烯分子模型;参考Huang等[2,3]报道的CuO/Cu2O界面模型,建立CuO/Cu2O(100)和CuO/ Cu2O(111)表面slab模型(图1)来分别模拟CuO/c-Cu2O和CuO/o-Cu2O;在提供的CuO/Cu2O(111)和CuO/Cu2O(100)表面(已结构优化)上初步确定可能的吸附位点并建立分子的吸附结构。
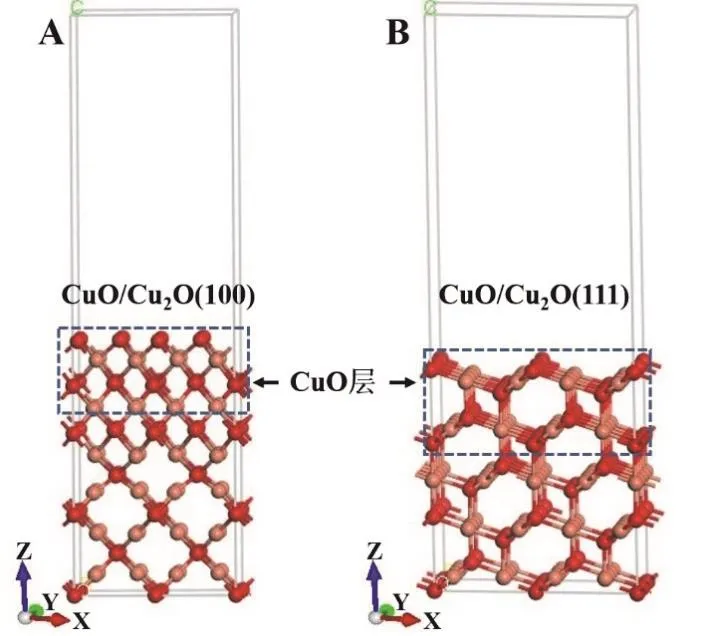
图1 通过Materials Studio 8.0软件建立CuO/Cu2O(100) (图A)和CuO/Cu2O(111) (图B)的表面结构模型
其中,在建立2 × 2的CuO/Cu2O(100)和CuO/Cu2O(111)的超胞模型时,选择x和y方向平行于表面而z方向垂直表面,同时在z方向选择18 Å的真空层以避免表面之间的相互作用力。在CuO/Cu2O(100)和CuO/Cu2O(111)结构中,分别共有13和15层原子层(z坐标相同的为同一层),其中在顶端的四层中均添加O原子构成Cu和O原子个数比为1 : 1的CuO层,同时在量化计算时,均需固定底端的原子,固定层数分别为5和6层。
1.4.3.2 量化计算及查看结果
使用SIESTA软件计算时,需准备输入文件(后缀名为.fdf)和赝势文件(后缀名为.psf)。准备输入文件时,从Materials Studio 8.0软件中导出小组分配好的结构文件(文件格式为.car),按格式生成计算所需要的坐标文件,设置计算方法和精度。在Xshell软件操作界面下向超算集群提交计算任务。计算结束后,从输出文件(OUT)中查看计算体系的能量值,计算分子在催化剂表面的吸附能。将得到的结构文件(后缀名为.STRUCT_OUT)转化成Materials Studio 8.0软件可识别的坐标文件格式,导入Materials Studio 8.0软件中,在可视化界面下查看结构信息。
2 结果与讨论
2.1 催化剂表征结果学习
2.1.1 Cu2O表征结果
c-Cu2O和o-Cu2O的FESEM与HRTEM表征结果如图2所示。其中,立方体的平均尺寸约为600 nm,八面体的平均尺寸约为400 nm。根据HRTEM图像图2C和图2F,c-Cu2O晶体的晶面间距为0.21 nm和0.21 nm,且晶面夹角约为90°,分别对应Cu2O的(020)面和(200)晶面(PDF卡片,No.78-2076)。o-Cu2O晶体的晶面间距为0.25 nm和0.25 nm,且晶面夹角约为71°,分别对应Cu2O的(111)面和(1-11)面(PDF卡片,No.78-2076)。以上可以说明制备得到的立方体和八面体两种Cu2O纳米晶主要暴露了(100)晶面和(111)晶面。

图2 (A) c-Cu2O的FESEM图;(B,C) c-Cu2O的HRTEM图;(D) o-Cu2O的FESEM图;(E,F) o-Cu2O的HRTEM图
2.1.2 CuO/Cu2O表征结果
对表面重构后的催化剂样品进行结构表征,结果如图3所示。由HRTEM图像可知,经过表面重构后,两组纳米晶表面产生一层约3 nm厚的薄膜。在XPS谱图中Cu 2p3/2的结合能位于933.2 eV,同时在940-945 eV出现卫星峰,证明Cu2O纳米晶表面的薄膜是被氧化产生的CuO薄膜。XRD图谱在29.6°,36.4°,42.3°,61.4°,73.5°出现的衍射峰,分别对应Cu2O的(110),(111),(200),(220),(311)晶面(PDF卡片,No.78-2076),说明CuO/c-Cu2O与CuO/o-Cu2O的体相结构仍然保持为Cu2O晶相。

图3 (A,B) CuO/c-Cu2O和CuO/o-Cu2O的HRTEM图,(C,D) CuO/c-Cu2O和CuO/o-Cu2O的XPS谱图和XRD谱图
2.2 活性测试
活性测试实验所得的结果如图4所示。CuO/c-Cu2O在270 °C开始显示活性,在300 °C时转化率为21.5%;而CuO/o-Cu2O在220 °C就开始显示活性,在300 °C转化率高达80.3%,说明CuO/o-Cu2O对乙烯的催化活性比CuO/c-Cu2O高。对Arrhenius曲线进行拟合,CuO/c-Cu2O和CuO/o-Cu2O催化乙烯氧化的表观活化能分别为117.0 ± 4.3 kJ·mol-1和77.7 ± 3.2 kJ·mol-1,两者的表观活化能差别很大,这说明两种催化剂在反应中的反应活性位点不同,并且遵循不同的反应机理。

图4 CuO/c-Cu2O和CuO/o-Cu2O催化乙烯氧化的转化率(A)和Arrhenius曲线(B)
转化率结果对于温度稳定时间、反应气流量十分敏感,且这两个变量引起误差无法消除,故学生在实验过程中需保证达到温度稳定时间并监控气体流量确保其状态稳定。在实验过程中,反应器到达设定温度后稳定15-20 min后开始测试,反应气流量需控制在20 mL·min-1。
2.3 量化结果分析
催化剂表面上未饱和配位的原子往往作为活性中心,在CuO/Cu2O(100)表面,暴露的表面未饱和配位的原子为单一的O2c;而在CuO/Cu2O(111)表面,表面暴露的未饱和配位的原子为Cu3c和O3c(图5(A)和(B))。计算乙烯在两个表面上可能的吸附结构,确定CuO/Cu2O(100)上的化学吸附位点是(O2c,O2c),而CuO/Cu2O(111)上有两种化学吸附的位点,一种是(Cu3c,Cu3c)位点,另一种是(O3c,O3c)位点(图5(C)-(E))。

图5 (A,B) CuO/Cu2O(100)和CuO/Cu2O(111)表面上乙烯的吸附位点;(C,D、E) 乙烯在CuO/Cu2O(100)和CuO/Cu2O(111)表面上稳定的吸附结构、吸附能(单位:eV)和C=C键长(单位:Å)
乙烯分子在CuO/Cu2O(100)表面上的吸附作用强,吸附能为-6.21 eV,其中,C=C键由原来的1.338 Å伸长至1.527 Å。而在CuO/Cu2O(111)表面上,乙烯分子在(Cu3c,Cu3c)和(O3c,O3c)位点上的吸附能分别为-3.11和-2.34 eV,对应的C=C键长分别为1.459和1.527 Å。可见,CuO/Cu2O(111)表面上,(Cu3c,Cu3c)位点更易吸附乙烯分子,而(O3c,O3c)对C=C键活化作用更强。比较乙烯分子在两个表面的吸附能,发现乙烯在CuO/Cu2O(100)表面吸附作用明显比在CuO/Cu2O(111)表面上高。然而,根据Balandin火山曲线规律,过强或者过弱的吸附强度都会导致催化剂的催化活性下降,具有适中的吸附强度对催化反应最有利[9]。通过文献学习可以推测在气固催化反应中,这个“适中”的吸附强度对应的吸附能在-1 - -2 eV[10]。所以,CuO/Cu2O(100)表面对乙烯的吸附能过高,可能会对反应过程产生抑制作用,而CuO/Cu2O(111)表面上乙烯的吸附能适中,有利于进一步反应。
2.4 综合分析
将活性测试实验结果与量化计算结果进行汇总,见表5。活性测试实验显示,CuO/o-Cu2O对乙烯氧化反应的催化活性比CuO/c-Cu2O要高,通过转化率数据计算得到两者的表观活化能差别很大,分别为77.7 ± 3.2 kJ·mol-1和117.0 ± 4.3 kJ·mol-1,说明这两种催化剂表面暴露的活性位点不同,且CuO/o-Cu2O表面的催化活性优于CuO/c-Cu2O表面。基于催化剂表面的实验表征结果,构建催化剂表面结构模型,能清晰直观地看到CuO/o-Cu2O的活性位点为(Cu3c,Cu3c)或(O3c,O3c),而CuO/c-Cu2O的活性位点为(O2c,O2c)。相比于CuO/c-Cu2O(100)表面,乙烯分子在CuO/o-Cu2O(111)表面的吸附能比较适中,有利于反应的进行和产物的脱附[9,10],从而对应并解释了活性测试实验结果。

表5 实验与量化计算结果
在实验中引入量化计算,不仅是让学生掌握一项实验技能,更是让学生具备实验结合理论、宏观联系微观的思维方式。然而,合理地将量化计算结果与实验结果相对应是分析的重点与难点。尤其对于多相催化这种复杂的反应体系,由于量子化学计算的一些局限性,一般的计算结果只能部分反映体系的真实状态。因此通过计算结果对实验结果给出一个恰当的解释是一个充满挑战的过程,往往需要查阅相关资料并结合体系自身的特点进行大量的讨论,而通过这一步的练习,充分锻炼了学生针对问题查阅文献、讨论思辨的能力,让学生在这个过程中加深对催化剂结构与性能之间关系的理解。
3 结语
催化剂的催化性能与其表面结构息息相关。基于科研工作者提出的模型催化理念,实验结合量化计算对表面催化过程进行研究,已成为研究催化剂表面科学的一种新方法。教学组将这一学科前沿研究方法引入到实验教学中,填补了催化剂表面结构与催化性能关系相关实验内容上的空白。通过文献调研、实验尝试和验证,教学组选择理化性能稳定,形貌规整,暴露特定晶面的CuO/Cu2O纳米晶作为模型催化剂,选择绿色安全的乙烯气体作为反应气,成功地将该体系引入实验教学。
教学中采取了实验与量化计算相结合的方式。实验中,学生进行活性测试并计算得到表观活化能。随后对乙烯分子在CuO/c-Cu2O和CuO/o-Cu2O表面吸附的结构进行量化计算,得到相应的结构信息和吸附能。量化计算结果与实验结果相互验证,互为解释,共同构建对催化剂表面结构与催化性能关系的合理的解释。通过对这个模型催化体系的学习,让学生对物化教材上的相关知识有更深刻的理解,掌握了催化活性测试的实验操作、表面结构模型的搭建方法和量化计算上机操作流程,锻炼了他们的文献调研能力,并激发高年级本科生的研究兴趣、培养综合实验技能,为他们能够顺利进入研究生学习阶段奠定良好基础。
