(2α,3α,5α,16β,17β)-2,3-环氧基-16-(1-吡咯)-17-羟基雄甾烷的合成及结构表征
2023-01-07马文静张梦敏翟李静宗雪梅马玉恒
马文静,张梦敏,翟李静,苗 霖,宗雪梅,王 栋,马玉恒
(1.滁州学院 材料与化学工程学院,安徽 滁州 239000; 2.江苏正大清江制药有限公司, 江苏 淮安 223001)
0 引言
罗库溴铵(Rocuronium Bromide)化学名为1-[(2β,3α,5α,16β,17β)-17-(乙酰氧基)-3-羟基-2-(4-吗啉基)雄甾-16-基]-1-丙烯基吡咯铵溴化物,其制剂商品名为Esmeron,由荷兰Organon公司于1994年6月和7月首次在美国和英国上市.本品是一种新型的非去极化甾醇肌松药,用于麻醉时的气管插管和手术时的肌肉松弛,具有起效快、持续时间短、恢复迅速、体内无蓄积等优点,是目前国际上应用最广泛的肌松药[1-3].
由于罗库溴铵所具备的独特优势及广泛的应用范围,引起了合成工作者和制药工作者的高度重视.目前常见的合成罗库溴铵有多条合成路线[4-12],这些路线均以(2α,3α,5α,16β,17β)-2,3-环氧基-16-(1-吡咯)-17-羟基雄甾烷(后简写为中间体1)为关键中间体,之后经过环氧化合物开环、17-羟基选择性乙酰化和吡咯环烯丙基化得到罗库溴铵.作为所有合成路线的关键中间体1,该化合物的结构确认及表征对罗库溴铵的质量至关重要(图1).因此,笔者利用1H-NMR、13C-NMR、DSC/TGA、粉末X射线衍射、单晶X射线衍射,对关键中间体进行结构表征和分析,并通过Hirshfeld表面计算从微观结构深入了解分子间作用力,为得到稳定性更好的罗库溴铵的原料药提供关键的结构信息,对罗库溴铵的临床安全用药具有重要的意义.
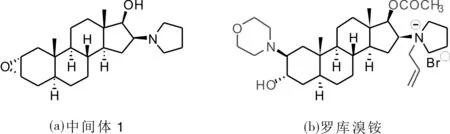
图1 中间体1与罗库溴铵结构
1 实验部分
1.1 仪器与试剂
1H-NMR 谱和13C-NMR 谱均用Brüker Advance 500 MHz 核磁共振仪测定(CDCl3为溶剂,TMS 为内标).高分辨质谱由 Applied Biosystems (ABI) Q-Star Elite MALDI-TOF 质谱仪测定.单晶结结构由Bruker Apex II CCD单晶衍射仪(Cu Kα,λ=0.071 073 nm)测定.X 射线粉末衍射谱用Bruker公司D8 ADVANCE测定.化合物2从表雄酮按照文献[10-11]方法,通过磺化消除、溴代、环氧化、哌啶取代等过程进行改进,实验室自制得到,其制备过程如图2所示.所有试剂均为市售分析纯,未进行进一步提纯.实验过程所用去离子水为自制超纯水.
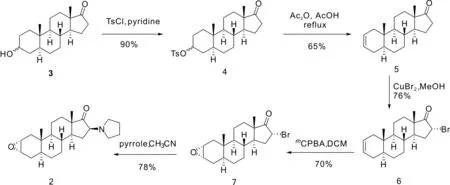
图2 化合物2的合成
1.2 中间体1的合成及单晶培养
在0 ℃的条件下,向化合物2 (1.80 g,5.0 mmol)的甲醇 (30 mL) 溶液中,缓慢加入NaBH4(0.76 g,20.0 mmol),在0 ℃条件下反应12 h后,缓慢加冷水30 mL,搅拌10 min,析出大量固体,过滤干燥,甲醇重结晶得到白色中间体1 (1.60 g,4.4 mmol),产率 89%(图3).1H-NMR (400 MHz,CDCl3)δ/10-6:3.36~3.14 (m,1H),3.12~3.10 (m,1H),2.95~2.88 (m,1H),2.76~2.74(m,2H),2.53~2.50(m,2H),1.95~1.89 (m,2H),1.88~1.81(m,2H),1.80~1.75 (m,3H),1.75~1.68(m,3H),1.68~1.64 (m,1H),1.57~1.46(m,2H),1.44~1.40 (m,1H),1.38~1.28(m,4H),1.27~1.22 (m,1H),1.21~1.05(m,2H),0.91~0.77(m,2H),0.76(d,3H),0.68(d,3H),0.67~0.60(m,1H);13C-NMR (100 MHz,CDCl3)δ=79.08,63.32,53.84,53.07,52.48,51.12,48.43,43.33,38.34,38.31,36.31,34.93,34.86,31.84,29.45,29.12,28.39,23.37,20.63,13.02,12.84.HRMS(ESI/[M+H]+) 360.2903,found 360.2900.
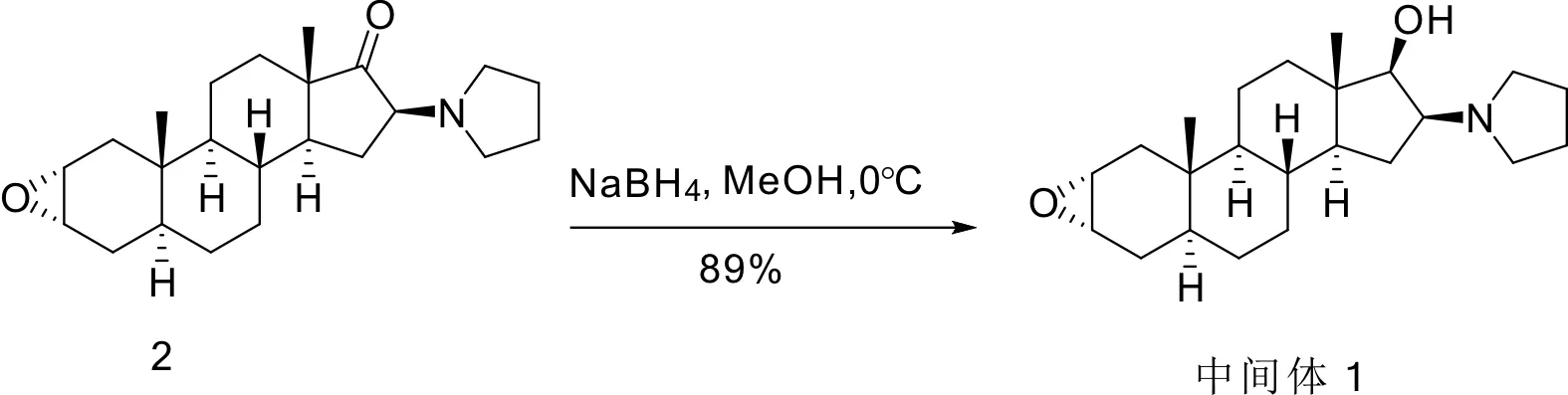
图3 中间体1的制备路线
单晶培养:分析天平称取100 mg研磨物,将其超声溶解在30 mL的甲醇溶液中,滤纸过滤,放置于室温下缓慢挥发,观察到5天后生成无色透明针状晶体.
2 结果与讨论
2.1 X射线粉末衍射PXRD
将适量研磨后的粉末制样后,采用德国Bruker公司D8 ADVANCE进行X射线粉末衍射的测定.实验条件:Cu (λ=0.154 06 nm),40 kV,40 mA,扫描步长为0.2°/s,记录2θ角从3°到40°的数据.所得数据用MDI-Jade软件处理.从图4可看出,原料的X射线粉末衍射图谱的特征衍射峰(2θ/(°))表现在5.96,11.91,15.55,16.22,17.02,17.90,18.08,18.60,20.97,21.71等衍射角处,尖锐的衍射峰表示该化合物状态结晶性良好.
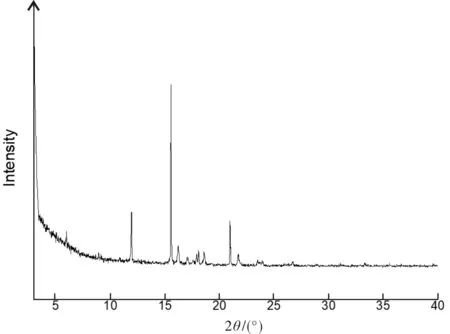
图4 中间体1的晶体的X射线粉末衍射图
2.2 单晶X射线衍射
选取合适尺寸的中间体1的单晶,置于Bruker Apex II CCD 单晶衍射仪上,在室温下采用Mo靶 Kα射线 (λ=0.071 073 nm),石墨单色器.晶体数据使用olex2软件进行解析,对于所有的非氢原子坐标和各向异性参数,用全矩阵最小二乘方法进行了修正[13-14].中间体1的晶体结构和结构修正数据如图5和表1所示.
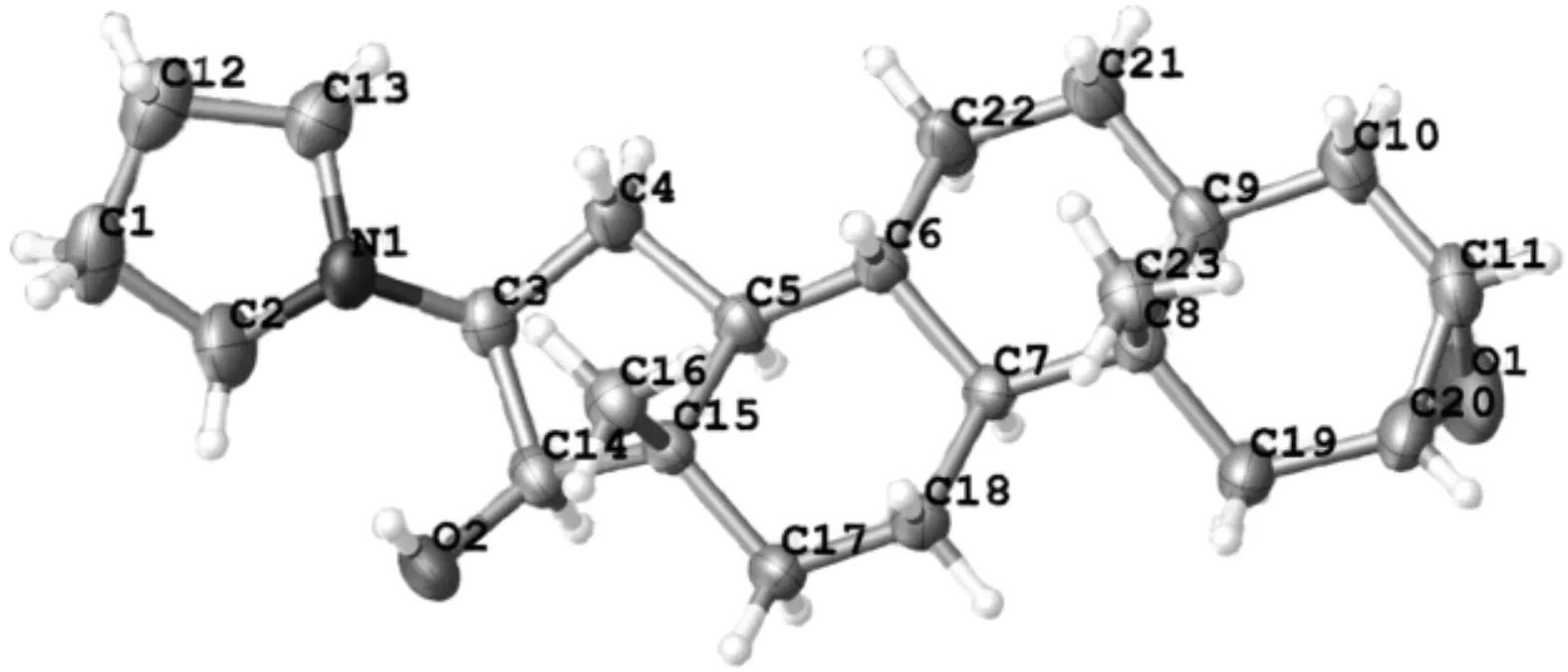
图5 中间体1的晶体结构
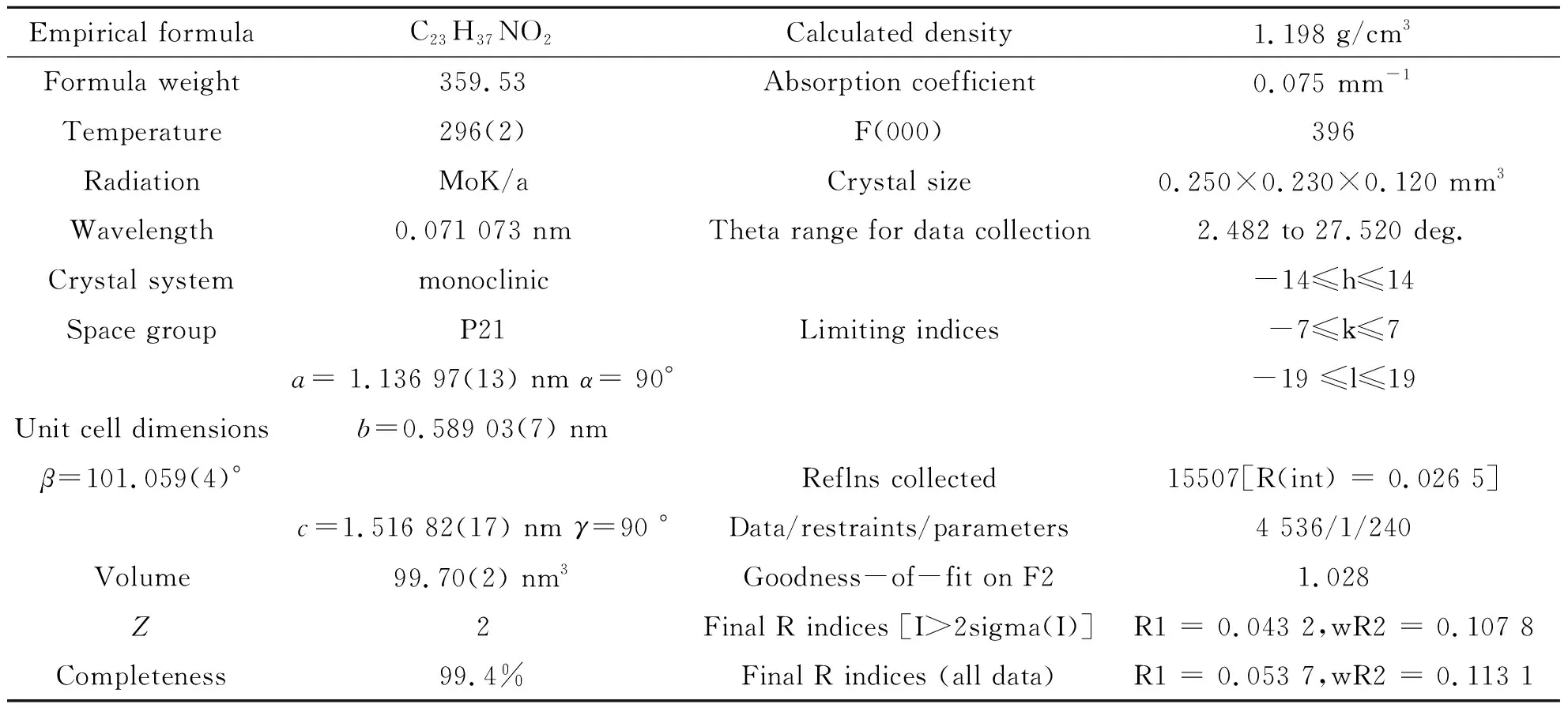
表1 中间体1晶体学数据和结构参数
中间体1的单晶为无色透明的块状晶体,呈现P21正交晶系.晶体结构如图5所示:C(14)—O(2)羟基朝上,其绝对立体构型为R-构型,可能因为旁边的四氢吡咯环和C(15)—C(16)处于六元环的朝上的直立键,所以还原H-是从面下进行,因此,生成的O(2)—H(35)朝上.晶体的部分键长和键角如表2、表3所示.

表2 中间体1结构的部分键长
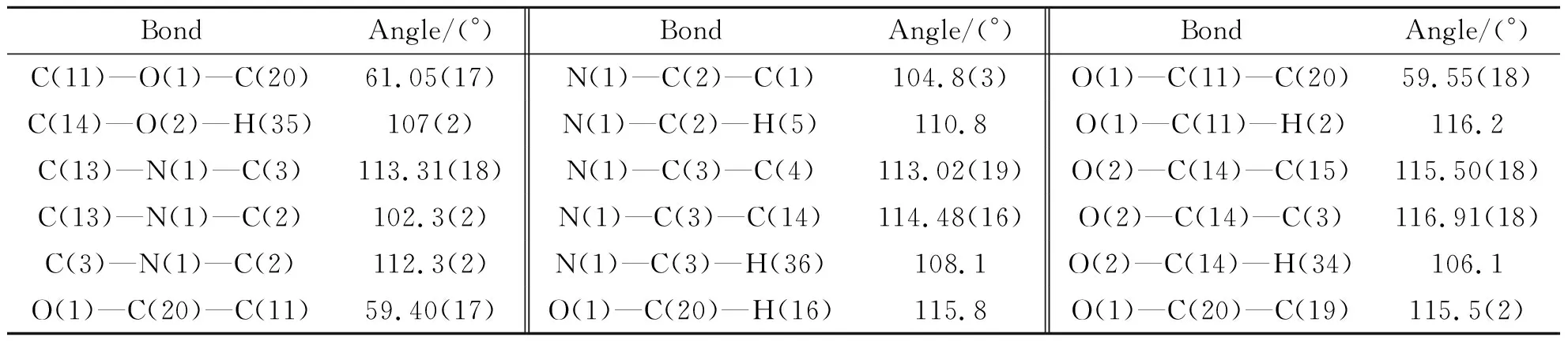
表3 中间体1结构的部分键角
为进一步研究分子间的作用情况,对晶体结构进行空间叠加,形成了一个二维片状结构,如图6所示.该二维结构有O(2)—H(35)…O(1) (x-1,y,z)和C(2)—H(5)…O(2)这两种氢键,如表4所示.O(2)—H(35)…O(1) 中H(35)…O(1)和O(2)…O(1) 之间的距离分别为0.218(3) nm和0.288 5(3) nm,角度为149(3)°,属于正常氢键范围.C(2)—H(5)…O(2)中H(5)…O(2)和C(2)…O(2)之间的距离分别为0.245 nm和0.302 0(3) nm,角度为117.1°.后者氢键距离更大,角度更小,相比于前者,氢键作用力较弱.这两种氢键扮演着桥梁的作用,将分子结构向外延伸,将分子间形成“之”字形的堆积结构,形成了一个沿着b轴的无限二维结构.
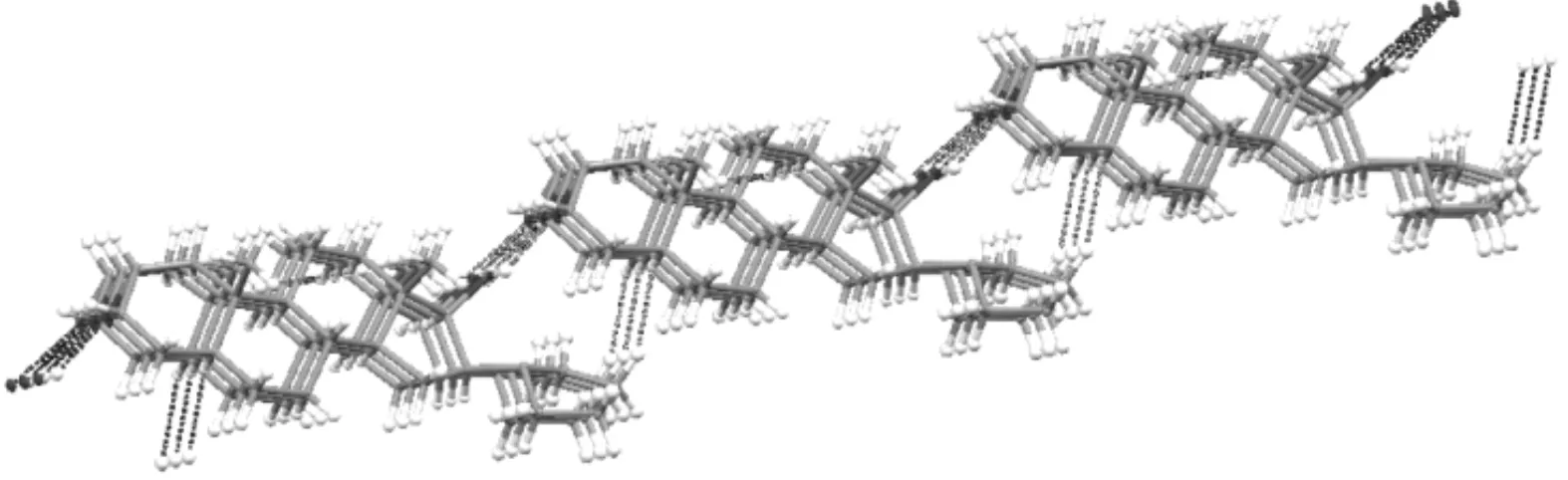
图6 沿b轴的二维片状结构
2.3 DSC/TGA分析
图7采用差示扫描量热法(DSC)和热重分析法(TGA)测定中间体1的稳定性.实验条件:样品称样量1.094 0 mg,升温速率10 ℃/min,氮气流量50 mL/min,温度范围为25 ℃~450 ℃.DSC图中呈现1个吸收峰,熔点160.45 ℃,结合TGA热重的分析可以得出:样品在151.44 ℃开始出现熔融现象,到168.94 ℃完全熔融;但这一过程,没有发生失重,之后样品开始分解,直至分解完成,总损失比重为93.460 8%.
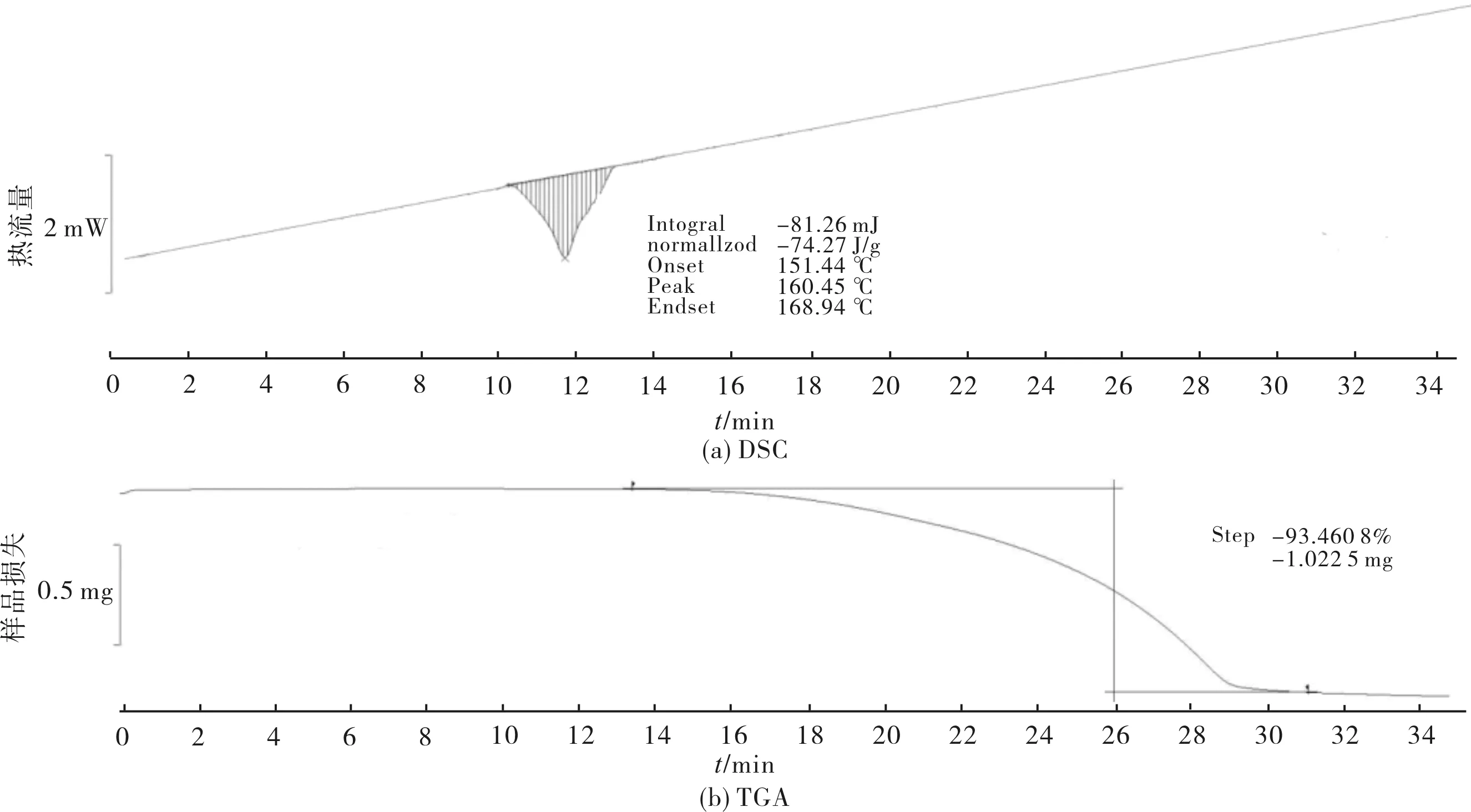
图7 中间体1的DSC和TGA图
2.4 Hirshfeld 表面分析
Hirshfeld 表面分析法是通过计算电子云密度揭示分子内、分子间键能直观多晶型结构研究方法[15-16].通过计算分子的Hirshfeld表面和二维指纹图谱,以不同的颜色表示原子作为氢键供体、受体的能力,白色相当于范德华半径的原子间距,蓝色代表比范德华力较长的距离,红色代表比范德华力较短的距离[17-18],可直观揭示分子间的相互作用,并能够区分晶胞中的不同作用力大小及所占比例.
中间体1的Hirshfeld 表面计算和指纹谱图分析是用Crystal Explorer 3.1 软件计算所得,如图8所示.图8(a)中1a、1b的斑点表示分子间的氢键连接,图8(a)中1a为羟基氢作为氢键供体O(2)—H(35)…O(1),斑点颜色深表示氢键作用力比较强;1b为碳氢键为供体C(2)—H(5)…O(2),颜色较浅表示氢键作用力较弱,这些信息与前面所列信息相互对应.图8(b)、8(c)为分子间作用指纹图谱.从二维指纹图谱中可以看出,H—H 作用力是全谱的主要构成部分,其中,O…H 间作用力占10.7%,C…H 间作用力仅占0.1%,与前面信息相对应.
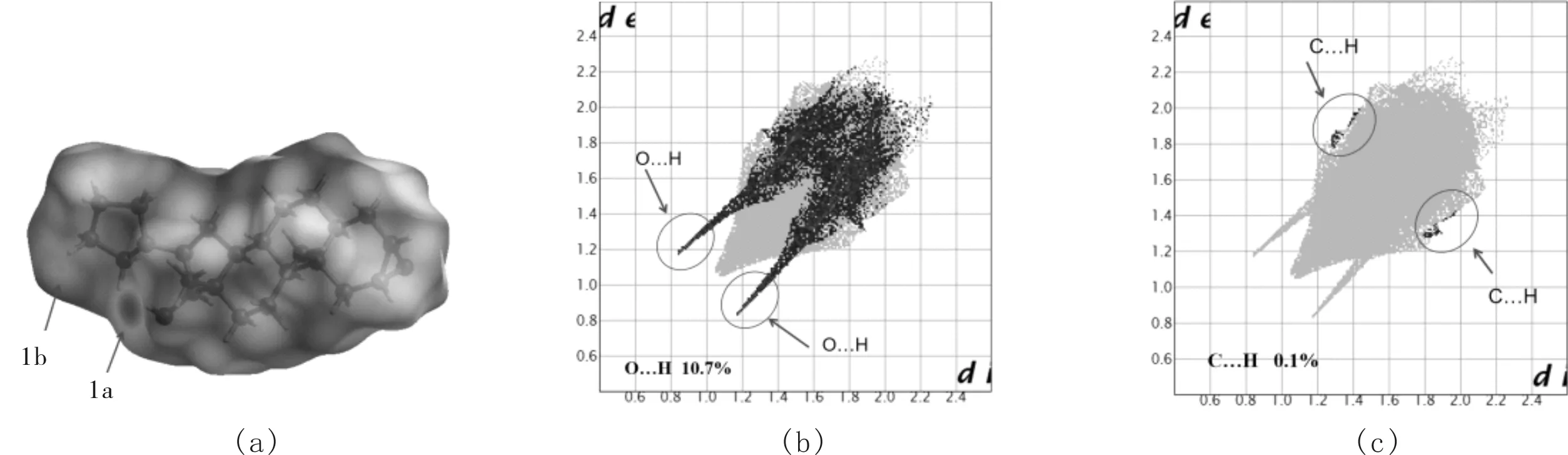
图8 中间体1的 Hirshfeld 表面分析及分子间作用力二维指纹图谱
3 结论
作为罗库溴铵合成的关键中间体,中间体1的全面结构解析对罗库溴铵的合成起着至关重要的作用.笔者采用硼氢化钠还原制备中间体1,并通过甲醇溶液缓慢挥发法成功制备其单晶结构,利用1H-NMR、13C-NMR、DSC/TGA、粉末X射线衍射、单晶X射线衍射对其进行表征和分析.单晶结构显示,晶格中存在O(2)—H(35)…O(1)(x-1,y,z)和C(2)—H(5)…O(2)这两种氢键的基础框架,其中,前者氢键较强,后者氢键作用较弱,这两种氢键结构使其在空间上折叠,形成二维片状结构.DSC/TGA显示,该化合物在151.44 ℃出现熔融现象.通过Hirshfeld表面分析及分子间作用力二维指纹图谱更进一步揭示分子间键能强弱,该结果与晶体分析结果一致.
