石墨相氮化碳的设计构筑对催化性能的影响
2023-01-07郑明明
王 珏,付 东,郑明明,阚 侃
(黑龙江省科学院高技术研究院,哈尔滨 150020)
石墨相氮化碳(g-C3N4,简称C3N4)是一种不含金属的有机半导体化合物,具有优异的热力学稳定性、化学稳定性和特殊的电子结构。C3N4中C、N原子以sp2杂化方式键合,其结构与石墨烯相似,是具有共轭π键的层状结构,凭借其特殊的性质在光解水制氢反应、氧还原反应、析氧反应、气敏传感器和电化学传感器等方面有着广泛的应用[1]。
体型C3N4可采用氰化铵、尿素、双氰胺、三聚氰胺、硫脲、三聚硫氰酸等含氮前驱体通过直接热聚合制备[2],但是直接热聚合获得的C3N4比表面积较低,活性位点较少,极大限制了C3N4的应用。而纳米结构设计、元素掺杂和异质结构是解决这一问题的最有效的方法[3]。
1 C3N4的纳米结构设计
纳米结构设计是一种改善体型C3N4缺陷的有效方法,通常包括零维点状、一维棒状、一维管状、二维片状和三维网络结构。与体型结构相比,零维点状、一维棒状、一维管状、二维片状和三维网络结构具有更高的比表面积,因此具有良好的吸附能力。同时也有研究者将纳米结构设计成多孔状,进一步提高材料的比表面积,使C3N4获得优异的吸附性能,提高催化效率。
零维点状的C3N4主要以C3N4量子点为主,通常与其他光催化纳米片材料复合,以提高基体材料的光催化性能。
Xue Lin[4]等将三聚氰胺放入马弗炉550℃煅烧4 h得到体型g-C3N4。将1 g体型g-C3N4在浓H2SO4和HNO3混合物中室温处理2 h,用去离子水稀释混合物,洗涤数次,将50 mg处理后的体型g-C3N4分散在30 mL氨水中,并转移至水热反应釜中,200℃反应24 h,冷却后超声6 h得到g-C3N4量子点(CNQDs)。采用光沉淀法,将Ag纳米颗粒与g-C3N4量子点沉积到Bi2MoO6纳米片上,得到CNQDs/Ag/Bi2MoO6纳米复合材料。CNQDs/Ag/Bi2MoO6纳米复合材料在可见光照射下对罗丹明B(Rh B)的降解性能提高约100%,其出色的光降解性能归因于CNQDs负载、高比表面积和三元复合体系中电子空穴对的有效分离所导致的可见光区的扩展。此外,自由基捕获试验证明空穴与O2·-自由基在Rh B的降解中起主要作用。
一维棒状C3N4凭借其优异的长径比具有很大的比表面积,使其可以产生更优异的光催化性能。
Hao Wang[5]等以三聚氰胺和三聚硫氰酸为原料,加入到DMF中,80℃搅拌形成超分子前驱体,经过热聚合得到S掺杂的C3N4。热聚合过程中,高熔点的三聚氰胺-三聚硫氰酸保留了有序结构,因此得到结晶度较好的多孔矩形棒状C3N4,如图1所示。但是这种C3N4的S掺杂含量较小,仅为0.14%,而少量的S掺杂就可增强可见光吸附,优化了光催化制氢的电子和带隙结构,其产氢效率较纯C3N4提高了11倍。

图1 多孔矩形棒状C3N4合成过程的示意图[5]Fig.1 Schematic diagram of the synthesis process of porous rectangular rod C3N4[5]
Yu Zhao[6]等采用水热法制备了纳米TiO2颗粒,采用简单的浸渍方法将纳米TiO2颗粒与棒状C3N4复合,得到了S-C3N4/TiO2复合材料。该复合材料在可见光照射下,表现出比纳米TiO2更好的光催化去除污染物的活性。由于电子-空穴对的有效迁移和转化、可见光吸收能力的提高及硫掺杂诱导的更大的比表面积,使得S-C3N4/TiO2复合材料的光催化活性增强。S的引入调控了C3N4的形貌,使超薄的g-C3N4纳米片组装呈棒状的S-C3N4/TiO2复合材料,这有利于提升S-C3N4/TiO2复合材料的催化性能。此外,通过捕获试验确定了S-C3N4/TiO2复合材料在催化反应中的主要活性物质为空穴与O2·-自由基,而不是·OH自由基。
一维管状结构在一维棒状结构的基础上进一步通过管状空腔增加了C3N4的比表面积,使其催化效果更好。一维管状的制备方法主要有软硬模板法[7-8]、低温处理法[9]、超分子自组装法[10]和离子液体辅助法[11]等。
Chengyin Liu[12]等将三聚氰胺与溴化铵用玛瑙研钵研磨混合,放入马弗炉中500℃煅烧,得到了一种角状中空介孔超薄C3N4管。其中,溴化铵起到了模板剂的作用,与三聚氰胺混合后生成了一种角状的中间体,热解后进一步分解得到角状多孔管,这种特殊的结构使C3N4具有高效的空间各向异性电荷分离能力,提高了管壳内外的电荷分离,使载流子密度较体型C3N4提高18倍,且提高了电荷的表面转移效率,因此光催化性能显著提高。Guifang Ge[13]等以三聚氰胺和三聚氰酸为前驱体,以氢氧化钾辅助水热处理得到棒状三聚氰胺-三聚氰酸超分子前驱体,KOH能够破坏超分子前驱体中的氢键,加速三聚氰胺的水解和氮缺陷的形成,经过高温热聚合得到C3N4管束结构。管束结构带来更高的比表面积,与KOH造成的丰富氮缺陷发挥协同作用,提高了光致载流子的可见光捕获能力和分离效率,使C3N4的光催化效率显著提高。Ruiru Zhao[14]等以正硅酸乙酯和氨水制备了纳米SiO2,并以其为造孔剂混合三聚氰胺,经过600℃热聚合得到含纳米孔的管状C3N4。管状结构赋予C3N4更高的比表面积,且纳米孔的边缘效应也使其对Rh B的降解效率更高。
二维片状C3N4通常分为自上而下和自下而上两种制备方式,其中自上而下的过程需要制备高质量的块状C3N4晶体,并将C3N4晶体剥离成片状的C3N4。由于聚合物单元链之间的氢键较弱,C3N4晶体平面原子结构会遭到破坏,从而产生材料缺陷。此外,超薄纳米片往往会因为π-π键相互作用而形成小型粉末或薄膜,降低离子的扩散速率及活性位点的活性,而自下而上的策略则可以有效避免这些弊端。
Dongni Liu[15]等采用低温退火法,以Co基ZIF-67为牺牲模板,制备了三维中空介孔Co3O4,并将其包裹在剥离的C3N4纳米片上,用于光催化氧化处理600 ppb的NO。在Co3O4和C3N4纳米片之间的p-n异质结形成了一个空间导电网络框架,使得可见光响应范围更广。Co3O4的中空介孔结构有助于NO的循环和吸附,较大的比表面积暴露了更多的活性位点。通过调节Co3O4前驱体的量,可使NO的降解效率达到57%。
Yuting Xiao[16]等提出了一种简便的分子自组装策略,通过三聚氰胺分子有序自组装及三聚氰胺水解产生三聚氰酸的过程,制备了一种棒状前驱体,这种前驱体具有0.315 nm的较大的层间距和-NH2、-OH等官能团,这种独特的性质使得乙醇、丙三醇等极性小分子可以很容易插入到层状前驱体中。醇分子插入后,通过热诱导剥离和热缩聚过程得到g-C3N4多孔薄片,过程如图2所示。多孔薄片状的g-C3N4具有超大的比表面积,催化位点暴露较多,有利于水接近反应活性位点,缩短了从内部到表面的迁移距离,有利于反应物和产物的扩散,显著改善了载流子的转移和分离,此外,氮空位引起的缺陷可作为分离中心,暂时捕获CB中的光诱导电子,更有效地激活分子氧,层状C3N4较体型C3N4的VB电位更大,因此层状C3N4的氧化能力更强,光催化制氢的活性更高。

图2 少层C3N4纳米片的制备示意图[16]Fig.2 Schematic diagram of the preparation process of few layer C3N4 nanosheets[16]
三维C3N4通常由低维C3N4组装而成,兼具了一维C3N4和二维C3N4的特性,通过低维C3N4构建的三维C3N4更具优异的性能。
Yibin Wei[9]等将三聚氰胺热聚合得到体型的C3N4,在乙醇中超声剥离得到C3N4纳米片,将C3N4纳米片在350℃热处理10 min,然后快速转移到冰水浴中形成C3N4纳米管。他们将C3N4纳米片与C3N4纳米管分别与石墨烯复合,得到了一维C3N4插层化合物和二维C3N4插层化合物。通过对比一维C3N4插层化合物和二维C3N4插层化合物发现,一维C3N4插层化合物对Rh B的吸附性更强,降解效率更高,石墨烯能够使C3N4的电子传输速率更快,因此Rh B的降解速率也更高。Mao Wu[17]等以双氰胺和植酸为原料,通过水热法得到了双氰胺溶液,冷却后将双氰胺溶液倒入液氮中迅速降温,冷冻干燥得到了一种一维/二维杂化体系,如图3所示。该体系同时结合了一维C3N4纳米管和二维C3N4纳米片,且植酸将P原子引入C3N4中形成P掺杂的C3N4。这种类海胆状的P掺杂一维/二维结构,可使C3N4的HER效率提高22.4倍。Young-Si Jun[18]等利用低维C3N4(如纳米颗粒、纳米管、纳米片)制备了三维宏观C3N4组件,同时利用三嗪环超分子合成了C3N4空心球。热聚合过程中,三聚氰胺与三聚氰酸之间的二维六角形组装使超分子演化为纳米片状结构。三聚氰酸、三聚硫氰酸等分子容易形成氢键网络,根据给-受体对和结晶溶剂的不同,可产生优先的晶体结构。“晶体工程”使C3N4的结构特性、缩聚过程和层间相互作用更容易调控,通过发挥不同微观结构的协同作用,使三维宏观组件的光催化性能提高。
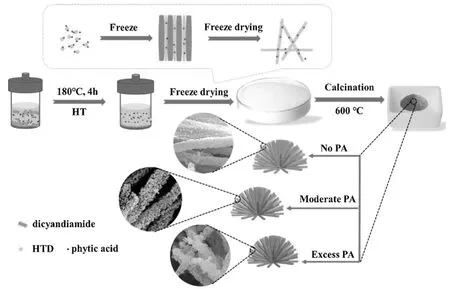
图3 三维多孔P掺杂C3N4纳米管的形成机理图[17]Fig.3 Formation mechanism diagram of three-dimensional porous P-doped C3N4 nanotubes[17]
2 杂原子掺杂C3N4
杂原子掺杂(如B、O、P、S等)可增加更多的结合位点,且外源元素可调节C3N4的能带结构和材料电导率,改善C3N4的表面性质。
Zhenyu Wang[19]等以三聚氰胺和硼酸为原料,通过180℃水热处理8 h,得到超分子前驱体,热聚合得到管状B掺杂的C3N4,如图4所示。管状B掺杂C3N4对NO的去除率高达30.4%,是体型C3N4的1.5倍。这种差异主要归因于管状B掺杂C3N4具有优良的光生电子-空穴对分离和载流子转移能力,通过超分子前驱体自组装,提出了一种同时控制外源原子掺杂和体系结构的策略。

图4 B掺杂C3N4纳米管前驱体在水热过程中的形态演化过程示意图[19]Fig.4 Schematic diagram of morphology evolution of B-doped C3N4 nanotube precursor in hydrothermal process[19]

3 C3N4异质结构的构建
两种不同的半导体材料相接触所形成的界面区域叫做异质结构,异质结构具有两种半导体各自都不能达到的优良的光电性能,构建异质结构有助于提高C3N4的性能。
Lijuan Ye[24]等将三聚氰胺与硫脲混合后充分研磨,平铺在坩埚底部,将ITO玻璃放在混合物上方,转移至马弗炉内,550℃加热4 h,得到了C3N4薄膜。将其放在钼酸钠溶液中,180℃水热24 h,用N2气流吹净表面得到MoS2/S-gC3N4异质结构,MoS2/S-gC3N4异质结构薄膜具有更优异的PEC性能,所产生的阳极光电流稳定达到1.2×10-4mA/cm2,是S掺杂C3N4薄膜的2倍。由于MoS2和S掺杂C3N4,之间形成了p-n异质结,因此PEC性能的提高主要是由于光致载流子的增加带来的协同效应。光致载流子的光吸收能力提高,电荷分离效率提高。
Tahir Muhmood[25]等以三聚氰胺为前驱体,通过热聚合法制备了C3N4,将红磷粉末加入到研细的C3N4中,在真空条件下600℃加热7 h,自然冷却后转移至氩气手套箱中,得到一种I型P/g-C3N4异质结,这种异质结的电子从C3N4向红磷迁移时,可使空穴因其边缘电位差而保持不动,对MTSM和RhB的降解时间短,这主要是由于C3N4中引入红磷可减少结构缺陷,提高电荷的分离能力,从而提高复合材料在可见光下的催化活性。
Huanyu Jin[26]等采用盐模板法制备了一种亲水性的二维MoN,以双氰胺为前驱体,通过带负电荷的MoN和带正电荷的双氰胺之间的静电吸引作用,使双氰胺沉积在MoN表面,经过氩气下冻干退火得到。这种二维g-C3N4@MoN异质结构具有双活性位点,能够促进界面上碱性HER的反应动力学。g-C3N4@MoN异质结构具有良好的电子性能和吸附性能,比大多数非贵金属催化剂具有更高效的电催化性能,而g-C3N4@MoN的高催化活性来自于MoN和g-C3N4分别对OH和H的高吸附性之间的协同作用,且每个活性位点对整体碱性HER活性的贡献很容易调整,因此二维C3N4@MoN是一种优异的HER催化材料。
4 结论与展望
通过材料设计,可使C3N4获得优异的光性能及电性能。由于独特的形貌和无金属π-共轭的低维共价有机骨架,使合成的C3N4在光解水制氢反应、氧还原反应、析氧反应、气敏传感器和电化学传感器等领域都有广泛的应用。但C3N4仍然存在几个难题:一是C3N4的可控制备及表面缺陷调控仍存在着一定的困难。二是纳米C3N4容易团聚,严重降低了二维形貌的独特结构优势。而对C3N4进行多种改性,发挥其之间的协同作用,是未来C3N4材料制备的主要方向,以实现针对特定目标的应用。
