团簇Fe3 Ni3稳定性及催化析氢性能
2022-12-30郑新喜方志刚侯欠欠
郑新喜,方志刚,秦 渝,侯欠欠,许 友
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
能源紧缺作为一个全球性的普遍状况,是当今世界需要迫切解决的重大问题之一,然而传统的化石能源发展潜力已经达到瓶颈,并且带来的环境污染、温室效应等已经给人类带来了巨大影响,摆脱化石能源的桎梏,研究和发展新型清洁能源,是当前世界能源发展的主流方向。其中,氢能以其热值高、无污染、利用形式多、安全性好等特点成为未来环保能源发展的大势所趋。由于其燃烧产物只有水,决定了其绿色能源的重要地位,目前氢能主要来源于光电催化反应制氢,但传统的铂碳催化剂制作成本较高、催化效率低,严重限制了制氢产业的发展。因此,寻找新型高效、绿色、低成本催化剂成为目前发展新型氢能的研究热点。
20世纪中叶发现的非晶态合金,由于其自身微观结构具有短程有序、长程无序的特点,使其在催化[1-2]、储氢[3-4]、磁性[5-6]等方面与传统的晶态合金相比,表现出更多令人欣喜的性能,该材料一经发现,便获得了广泛关注和研究,如今已经取得众多研究成果。其中Fe基[7]、Ni基[8-10]由于其成本较低、安全无毒、物理化学性能优越,符合绿色化学发展趋势等特点,成为目前非晶合金研究的热点内容,如Fe-Cr[11]、Fe-Co[12-13]、Fe-Cr-Ni[14-15]等 体 系 已 取 得众多显著的研究成果。WANG[16]的研究团队在非晶合金Co3MoS的密度泛函领域取得进展,证明了通过密度泛函计算研究催化析氢性能的可行性;QIN等[17]研究发现非晶合金Co3NiB在催化析氢方面具有优良的催化活性;ZHENG[18]的研究发现二元体系非晶态Fe-Ni合金在MOF结构材料中的电解水OER性能表现突出;CHEN等[19]发现Fe-Ni合金可以通过熔融纺丝法制备成表面质量良好的完全非晶结构,这为Fe-Ni非晶合金的制备提供了一个研究方向。
尽管非晶态Fe-Ni合金具有众多优异性能与良好的发展前景,但关于其催化析氢等方面的研究目前鲜有报道。因此本研究选择二元非晶态Fe-Ni合金体系进行研究,根据YANG等[20]的研究成果设计了团簇Fe3Ni3结构模型,然后利用密度泛函进行优化计算,从热力学能量与前线轨道理论两个方面针对对团簇Fe3Ni3的催化析氢性质展开研究,期望能够对该体系的催化析氢材料制备与生产提供理论上的指导,填补该领域的空白。
1 优化构型及其能量
本研究依据拓扑学原理[21],首先将团簇Fe3Ni3的初始经典空间构型尽可能设计出来,例如常见的平面构型、三棱锥型、四角双锥型等。依据密度泛函理论[22-23](Density functional theory,DFT),在B3LYP/Lan12dz量子化学水平下,将团簇Fe3Ni3的典型初始空间构型采用Gaussian09软件包和Multiwfn软件包[24]对其初始构型进行全频率验证和几何构型优化以及数据处理。其中Fe、Ni原子采用HAY等[25]的含相对论校正的有效核电势价电子从头计算基组,即18-eECP的双ξ基组(3s,3p,3d/2s,2p,2d)。
将优化后的数据进行分析处理,首先排除不稳定的含虚频构型以及同重态下能量较高的相同构型后,最后得到9种优化构型(其中a~e为三重态构型,f~i为单重态构型)。
由于构型1(3)的校正能最低,因此将其作为基准,分别计算出其他8种构型的相对于构型1(3)的能量,并将其计算结果标注于构型下方,其中序号上标括号内的数字为该构型的自旋重态,如图1所示。观察图中各优化构型的空间结构可以发现,在团簇Fe3Ni3的9种优化构型中,有7个为四角双锥型,说明四角双锥空间结构是团簇Fe3Ni3的优势空间构型,为实际的生产制备提供依据。同时发现,三重态构型均为四角双锥型,并且该重态构型能量均小于单重态构型,说明其三重态构型热力学稳定性均比单重态构型更加优异,其中构型1(3)的热力学稳定性最好,构型4(1)的稳定性最差。
2 团簇Fe3 Ni3析氢性能分析
2.1 团簇Fe3 Ni3析氢反应机理研究
在QIN等[17]的研究方法上,设计了团簇Fe3Ni3(下文均使用M表示)作为析氢反应模型,利用该模型可以将催化光电解水析氢时的反应机理[26]整体分为以下两步:
第1步:
第2步:第二步又有两种反应途径,具体步骤如下:
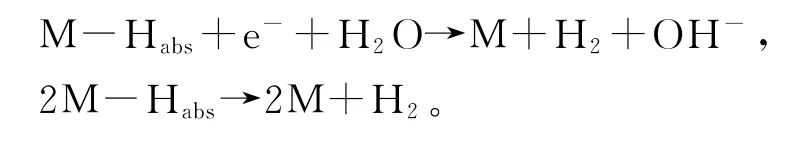
2.2 团簇Fe3 Ni3吸附H原子研究
2.2.1 前线轨道分析
利用前线轨道理论,可以针对化学反应中的电子跃迁问题进行理论分析,研究反应过程中的电子流向问题以及跃迁难易程度。该理论对分子轨道进行了区分和定义,其中HOMO轨道为电子占据的能量最高轨道,LUMO轨道为没有被电子占据的能量最低轨道,二者是决定一个体系是否发生化学反应的关键,对此进行研究可以探寻在团簇Fe3Ni3的模拟析氢反应中哪些轨道更容易完成第一步反应。
由于模拟析氢反应机理的第一步是电子从团簇Fe3Ni3的HOMO轨道跃迁至水分子的LUMO轨道,形成M-H模型(M为团簇Fe3Ni3)。为便于分析,依据计算结果,将各优化构型的α、β电子的HOMO轨道图以及H2O的LUMO轨道图绘制成图2。其中,白色离域部分表示轨道波函数的正相位,而黑色则表示轨道波函数的负相位,二者共同组成了电子在前线轨道中出现时的离域空间,也就是该构型在参与反应过程中活性最高的部分。
首先观察图2中的H2O-LUMO图,可以明显看出水分子周围均是波函数为负相位的黑色区域,由于轨道正负相位相同才更容易发生电子跃迁,进而发生反应,因此只要观察各构型α和β电子的HOMO轨道图中代表负相位的黑色区域即可。

图2 团簇Fe3Ni3的α、β电子的HOMO轨道图与水分子的LUMO轨道图Fig.2 HOMO orbital diagram ofαandβelectrons and LUMO orbital diagram of water molecule in Fe3 Ni3 cluster
从整体来看,9种优化构型各自的α-HOMO和β-HOMO的轨道图中,代表负相位的黑色区域与代表正相位的白色区域形状和大小均不相同,说明9种构型与水分子之间发生反应的难易程度也不一样,这也证明了选取的9种优化构型都具有代表性。其中三重态构型中的1(3)、2(3)和4(3)这3种构型,各自的α-HOMO轨道图中黑色部分均大于β-HOMO轨道图中的黑色部分,即α-HOMO轨道图中相位为负的离域空间更大,因此更容易与外来水分子LUMO轨道相位为负的离域空间发生反应,而构型3(3)与构型5(3)的α-HOMO、β-HOMO轨道图中黑色部分大小相似,这两种构型并没有表现出明显的的反应倾向。而4种单重态构型中,构型3(1)的HOMO轨道图中黑色部分最大,电子更容易完成向水分子的跃迁,构型1(1)与构型2(1)次之,构型4(1)的HOMO轨道图黑色部分最小,与水分子LUMO轨道中相位为负的部分进行重叠,电子发生跃迁,使构型吸附上氢原子的难度最大。
综上分析,团簇Fe3Ni3三重态构型中,各优化构型的α-HOMO轨道的负相位离域空间更易与外来的水分子负相位离域空间发生重叠,进而电子发生跃迁完成反应机理中的第一步—吸附氢原子;在单重态构型中,构型3(1)的HOMO轨道反应倾向最强。
2.2.2 团簇Fe3Ni3与水分子间的轨道能级差
团簇Fe3Ni3的HOMO图与水分子LUMO图仅能表示电子在两种轨道中出现的离域空间,而不能完全反映出电子的具体离散情况,仅仅以此判断该团簇析氢反应的难易程度存在局限性。为弥补该局限性,接下来通过轨道能级差进一步分析。
前线轨道理论指出,参与化学反应过程中的各个分子间具有较小的前线轨道能级差,电子才能在HOMO轨道与LUMO之间发生跃迁。同时模拟析氢反应机理中发现,当团簇Fe3Ni3的HOMO轨道与水分子的LUMO轨道能级差小于579 kJ·mol-1[27]时更有利于促进电子发生跃迁,使其更好地完成析氢反应第一步。根据计算结果,我们将团簇Fe3Ni3的HOMO能级(EHOMO)、水分子的LUMO能级(ELUMO),以及两者之间的能级差均列于表1当中。
由表1可以直接看出,团簇Fe3Ni3各优化构型HOMO能级与水分子LUMO能级差(ΔE)均小于上文中所要求的579 kJ·mol-1,也就是说9种优化构型在析氢反应第一步中均具有较好的反应活性。
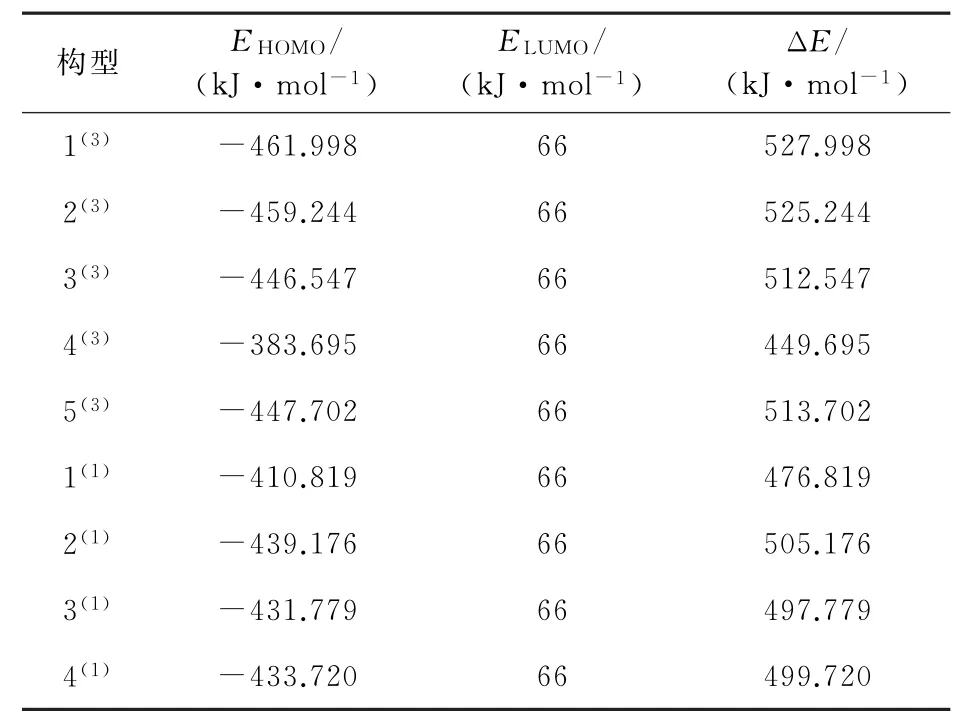
表1 团簇Fe3 Ni3的HOMO能级与H2 O的LUMO能级以及二者的轨道能级差Table 1 HOMO energy level of cluster Fe3 Ni3 and the LUMO energy level of H 2 O and the orbital energy level difference between them
具体来看,构型1(3)的轨道能级差最大(ΔE=527.998 kJ·mol-1),电子从该构型的HOMO轨道跃迁到水分子的LUMO轨道所需的能量最大,因此难度最高,其活性最弱;而构型4(3)的轨道能级差最小(ΔE=449.695 kJ·mol-1),跃迁所需能量最小,跃迁更容易发生。这说明团簇Fe3Ni3各优化构型模拟析氢反应第一步中,构型1(3)在吸附氢原子的难度最大,而构型4(3)的反应难度最小,最有利于发生氢原子吸附反应。将第一步吸附氢原子过程中各优化构型发生反应的活性从大到小排序为:4(3)>1(1)>3(1)>4(1)>2(1)>3(3)>5(3)>2(3)>1(3)。
2.3 (Fe3 Ni3)-H解吸过程
2.3.1 (Fe3Ni3)-H与H2O的前线轨道分析
前文2.2部分从前线轨道理论对团簇Fe3Ni3各优化构型与水分子之间的催化析氢中的第一步吸附氢原子进行分析,得到9种构型在完成第一步反应时的活性比较,同时完成反应第一步时,团簇模型吸附氢原子与水分子形成Fe3Ni3-H(M-H)结构,如图3所示。由图3可以明显看出,团簇M-H结构中除构型1(1)外,其余所有构型的的氢原子都与Fe原子进行结合,说明Fe原子是该团簇模型析氢反应过程中的主要吸附活性位点。
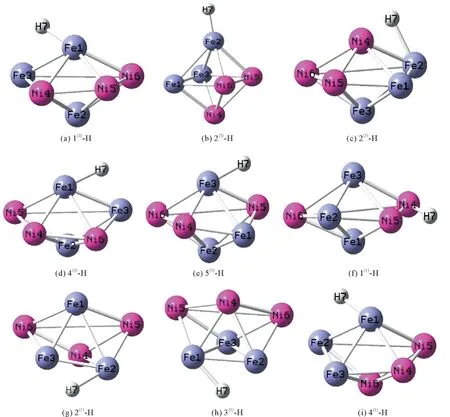
图3 (Fe3 Ni3)-H空间构型图Fig.3 (Fe3 Ni3)-H spatial configuration diagram
当析氢反应中第一步吸附氢原子完成后,M-H模型将会继续与另一个水分子发生反应并吸附第二个氢原子,然后两个氢原子相结合形成H2分子,从团簇模型上脱离下来,完成电子由水分子到M-H的转移,团簇恢复到反应发生之前的结构,析氢反应第二步结束。一轮催化析氢反应结束后,团簇将结合新的水分子开始下一轮反应。
第二步中的M-H解吸过程的难易程度直接决定了该步骤的反应速率,因此接下来对M-H模型的HOMO轨道与水分子的LUMO轨道能级差进行研究,进而判断各构型在进行第二步反应时的活性大小。将9种M-H模型的HOMO轨道与水分子LUMO轨道的能级差列入表2当中,根据表中数据分析,9种M-H模型的轨道能级差均小于579 kJ·mol-1,因此9种M-H模型均能完成析氢反应中的第二步反应,同时也印证了引言中Fe-Ni二元体系具有良好的催化析氢这一结论。虽然2.2部分中构型4(3)的轨道能级差最小,在析氢反应第一步的活性最大,但是其对应的M-H模型轨道能级差最大,在第二步反应过程中H2脱离的难度最大;2(1)-H模型的轨道能级差最小,且相较于吸附氢原子前构型2(1)的轨道能级差有所下降,其反应活性有所增加。因此综合分析可知:在第二步解吸过程中,模型2(1)-H的反应活性最强,构型4(3)-H的反应活性最差。将第二步解吸过程中M-H模型按照与H2O反应活性大小进行排序:2(1)-H>4(1)-H>1(1)-H>3(3)-H>1(3)-H>3(1)-H>5(3)-H>2(3)-H>4(3)-H。
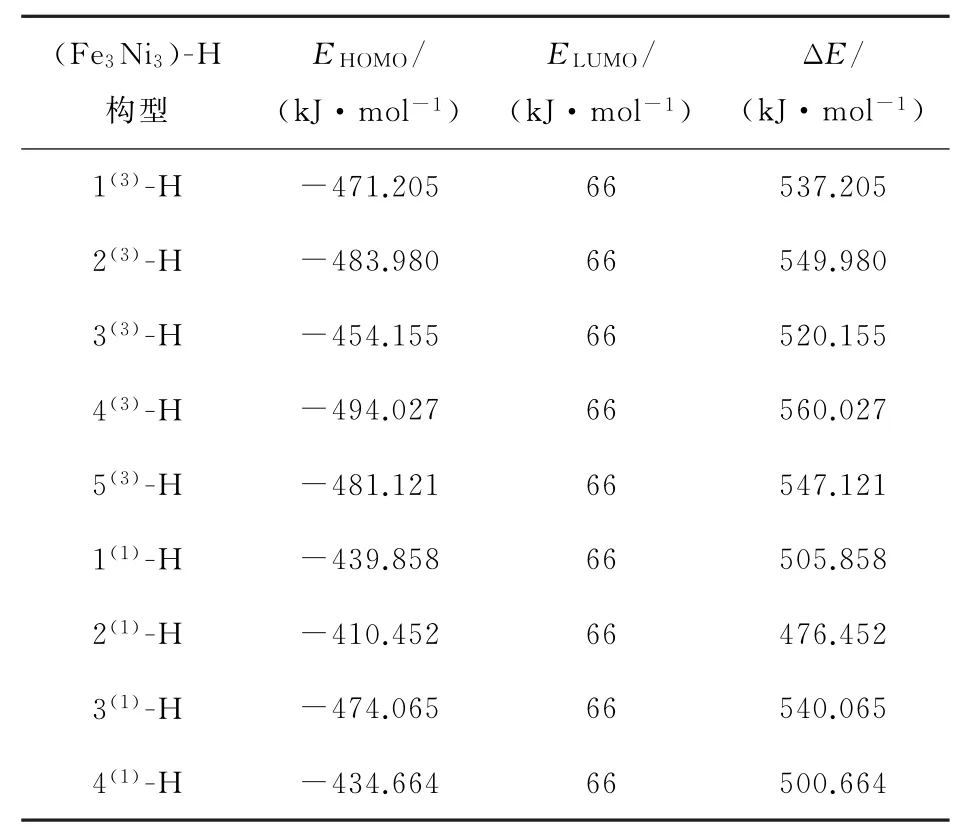
表2 (Fe3 Ni3)-H构型HOMO轨道与水分子LUMO轨道以及二者的能级差Table 2 (Fe3 Ni3)-H configuration HOMO orbital and water molecule LUMO orbital and their energy level difference
结合前文2.2.2部分的结论综合分析,团簇Fe3Ni3在参与电解水催化析氢反应过程中9种优化构型均具有良好的催化活性,其中构型1(1)在催化析氢反应过程中,无论是第一步还是第二步反应都具有较好的反应活性,因此在整个催化反应过程中活性最好,而构型2(3)与之相反,两步反应中活性均较差,整体反应活性最差。
2.3.2 (Fe3Ni3)-H分子轨道图分析
由于析氢反应第二步中,Fe3Ni3-H模型需要与另一个H2O进行结合,进而脱去H2分子,通过模型(Fe3Ni3)-H分子轨道图对各模型的α-HOMO、β-HOMO轨道在第二步反应过程中的贡献情况进行分析,(该部分分析方法与2.2.1部分分析方法一致),以便研究其具体反映机理。图4中模型的黑色部分(负相位离域空间)越大,则说明该模型越容易与第二个水分子发生反应,进而更好地完成第二步析氢反应。
对图4进行分析,整体来看各M-H模型的α与β电子的HOMO轨道图中黑色和白色两部分空间形状和大小分布均不相同,其轨道波函数正负相位的重合程度也不相同,这也与2.2部分中的情况完全一致,说明各M-H模型与另一个水分子之间发生反应的难易程度也有所差异。对模型Fe3Ni3-H的HOMO图进行具体分析,发现模型2(3)-H、3(3)-H与模型1(1)-H、2(1)-H、3(1)-H和4(1)-H的α-HOMO轨道图中黑色部分(负相位离域空间)大于各自的β-HOMO轨道图中黑色部分,说明上述模型的α-HOMO轨道更易与另一个水分子发生第二步析氢反应;而模型1(3)-H与模型5(3)-H的α-HOMO、β-HOMO轨道图中黑色部分大小相似,说明这两种模型与另一个水分子发生反应时α-HOMO、β-HOMO两个轨道活性相近;而模型4(3)-H的α-HOMO轨道图中黑色部分小于β-HOMO轨道图中黑色部分,说明该模型在第二步反应中β-HOMO轨道更具有反应倾向。

图4 模型Fe3 Ni3-H的α、β电子的HOMO轨道图与水分子的LUMO轨道图Fig.4 HOMO orbital diagram ofαandβelectrons of model Fe3 Ni3-H and the LUMO orbital diagram of water molecule
整体而言,单重态构型形成的M-H模型在与另一个水分子完成第二步析氢反应时,α-HOMO轨道为主要贡献者,更易与另一个水分子的负离域空间进行重叠;而三重态构型形成的M-H模型中,仅模型2(3)-H与模型3(3)-H的α-HOMO轨道在催化反应中起主要贡献作用,模型1(3)-H与模型5(3)-H的α-HOMO、β-HOMO轨道贡献相似,模型4(3)-H的β-HOMO轨道贡献大于其α-HOMO轨道贡献。
3 结 论
1)团簇Fe3Ni3共有9种优化稳定构型,其中三重态构型5种,单重态构型4种,三重态优化构型的稳定性均优于单重态构型。
2)团簇Fe3Ni3所有构型均能参与催化析氢反应的两个步骤,其中Fe原子是催化的主要活性位点。
3)团簇Fe3Ni3在催化水析氢第一步反应过程中,三重态构型中的α-HOMO轨道负相位部分更容易与水分子LUMO轨道中负相位部分发生重叠,电子发生跃迁,从而完成第一步吸附氢原子的反应。其中构型4(3)反应活性最大,构型1(1)次之;第二步反应过程中单重态构型形成的M-H模型中α-HOMO轨道贡献最大,更易与另一个水分子结合完成第二步析氢反应,而三重态构型形成的M-H模型中,仅模型2(3)-H与模型3(3)-H的α-HOMO轨道在催化反应中起主要贡献作用;模型2(1)-H解吸H2的能力最强,反应活性最好,模型4(3)-H的反应活性最差。
综合催化析氢两步反应分析:构型1(1)与构型2(1)在所有构型中的反应活性最好。因此在未来的实验制备中可以参考这两种构型的空间结构,制备催化活性优良的非晶催化剂。
