基于中国人Lynch 综合征临床标准的家族性结直肠癌胚系突变谱多中心研究*
2022-12-29杨梦园朱丽珍张鼎吴斌顾艳宏吴涛张敬东邱萌潘杰袁瑛
杨梦园 朱丽珍 张鼎 吴斌 顾艳宏 吴涛 张敬东 邱萌 潘杰 袁瑛
约1/3 的结直肠癌(colorectal cancer,CRC)患者有肿瘤家族史,但是目前仅5%左右结直肠癌可检测出胚系致病性突变,即确诊为遗传性结直肠癌,其中以Lynch 综合征最为常见。Lynch 综合征是一种常染色体显性遗传的恶性肿瘤综合征,表现为罹患结直肠癌、胃癌、子宫内膜癌、小肠癌、输尿管或肾盂癌、卵巢癌和肝胆系统肿瘤等恶性肿瘤风险显著增高,是由5 个DNA 错配修复(mismatch repair,MMR)基因(MLH1、MSH2、MSH6、PMS2和EPCAM)突变引起,以MMR 功能缺陷(deficient MMR,dMMR)为主要分子病理特征[1]。Lynch 综合征的确诊依赖于胚系致病突变的检出,然而推荐所有结直肠癌患者接受Lynch综合征相关胚系测序有悖于卫生经济学目标,因此通常采用肿瘤家族史对结直肠癌患者进行初筛,再建议其中的高风险人群进行后续胚系检测与遗传咨询。
目前国际上通用的Lynch 综合征临床诊断标准和临床筛查标准分别为Amsterdam Ⅱ标准和改良的Bethesda 标准[2]。然而,国际上的通用标准并不适合规模日益小型化的家庭。基于Yuan 等[3]的研究成果和中国家系特点,2003 年全国遗传性大肠癌协作组提出中国人Lynch 综合征临床标准[4]:家系中至少有2例组织病理学明确诊断的CRC 患者,其中2 例为父母与子女或同胞兄弟姐妹的关系(一级血缘亲属),且符合以下任一条件:1)至少1 例为多发性CRC 患者(包括腺瘤);2)至少1 例CRC 发病年龄<50 岁;3)家系中至少1 例患Lynch 综合征相关肠外恶性肿瘤(包括胃癌、子宫内膜癌、小肠癌、输尿管和肾盂癌、卵巢癌和肝胆系统癌)。
根据既往研究报道,仅一半左右符合AmsterdamⅡ标准的家系和65%符合改良的Bethesda 标准的微卫星高度不稳定(microsatellite instability-high,MSI-H)家系能检测出MMR 基因突变[5-6]。而符合中国人Lynch 综合征临床标准的患者中近30%可最终确诊为Lynch 综合征[3,7]。但是,这些中国人Lynch 综合征临床标准相关研究大多开展于2000 年左右,当时仍采用一代测序作为主要的基因检测手段。如今,二代测序技术在临床中的应用日益广泛,实现了利用有限的生物样本,高通量、高效率检测多个基因,也为Lynch 综合征的临床诊治带来了新的契机。因此,前期本课题组开展了一项基于中国人Lynch 综合征临床标准的人群研究,利用一个包含61 个已报道与遗传性消化道肿瘤相关基因的二代测序平台,对入组患者进行胚系突变检测,旨在获得二代测序时代下符合中国人Lynch 综合征临床标准的家族性CRC 患者最终确诊Lynch 综合征的比例,同时建立起中国人群的家族性CRC 胚系突变谱。
1 材料与方法
1.1 材料
1.1.1 临床资料 基于中国抗癌协会大肠癌专委会遗传学组和中国医师协会结直肠肿瘤专业委员会遗传专委会平台,本课题组织了一项按照中国人Lynch 综合征临床标准使用二代测序平台诊断Lynch 综合征的开放性、多中心研究(Clinicaltrials.gov 注册号:NCT03046849),共收集了85 例就诊于国内7 家医院(浙江大学医学院附属第二医院、北京协和医院、江苏省人民医院、北京大学第一医院、辽宁省肿瘤医院、四川大学华西医院和温州市中心医院)符合中国Lynch 综合征临床标准的CRC 患者的临床信息,并对其进行外周血采集。这85 例患者相互间无血缘关系。此外,对于同意提供其他家系成员样本的家系,同时采集这些家系成员的外周血、唾液等样本。每位提供样本及临床信息的患者和家系成员均同意并签署《遗传性结直肠癌胚系突变研究知情同意书》。本研究经浙江大学医学院附属第二医院伦理委员会审核同意后实施(伦理批准号:2017-011)。
1.1.2 提取研究对象基因组DNA 应用 DNeasy Blood &Tissue Kit(69506,Qiagen 公司,美国),由QIAcube 核酸自动提取仪(Qiagen 公司,美国)完成患者及家系成员的外周血DNA 提取。应用口腔拭子DNA 提取试剂盒(天根公司,中国北京)提取家系成员的唾液DNA。应用QIAamp DNA FFPE Tissue Kit(56404,Qiagen 公司,美国)试剂盒提取石蜡包埋的肿瘤组织DNA。
1.2 方法
1.2.1 二代测序 采用深圳华大基因股份有限公司的BGISEQ500 遗传性肿瘤组件(深圳华大基因股份有限公司,中国),检测61 个与遗传性消化道肿瘤相关的基因:APC,ATM,AXIN2,BARD1,BMPR1A,BRCA1,BRCA2,BRIP1,CDC73,CDH1,CDK4,CDKN1B,CDKN2A,CHEK2,EPCAM,EXT1,EXT2,FH,FLCN,MAX,MEN1,MET,MLH1,MLH3,MRE11A,MSH2,MSH6,MUTYH,NBN,NF1,NF2,NTRK1,PALB2,PMS1,PMS2,PTEN,RAD50,RAD51C,RB1,RET,SDHAF2,SDHB,SDHC,SDHD,SMAD4,STK11,TMEM127,TP53,VHL,RAD51D,SMARCA4,HOXB13,BLM,GALNT12,MSH3,NTHL1,POLD1,POLE,KIT,PDGFRA,SDHA。通过构建相应二代测序文库、上机测序(BGISEQ500 测序仪,深圳华大基因股份有限公司,中国),对每个基因外显子及其邻近±10 个碱基的内含子区变异(包括点突变和20 个碱基以内的缺失插入突变)、拷贝数变异情况进行分析。根据相对于人群数据库(千人基因组数据库和 ExAC 数据库)数据和本地数据集确定的最小等位基因频率(minor allele frequency,MAF),保留MAF 小于0.1%的突变。根据美国医学遗传学与基因组学学会(American college of medical genetics and genomics,ACMG)指南对胚系变异进行分类,包括良性、疑似良性、临床意义不明、疑似致病和已知致病五类[8]。
1.2.2 多重荧光多聚酶链反应(polymerase chain reaction,PCR)毛细管电泳法 构建PCR 扩增体系,5个微卫星位点对应PCR 引物如下:NR-21(F:GAGT CGCTGGCACAGTTCTA;R:CTGGTCACTCGCGT TTACAA)、NR-22(F:GGATAATCGAGGCTTGTCA AG;R:GCCCAAGACAAAACTTCCAG)、NR-24(F:GTGTCTTGCTGAATTTTACCTCCTGAC;R:ATTG TGCCATTGCATTCCAA)、BAT-25(F:CTCGCCTCC AAGAATGTAAGT;R:TCTGCATTTTAACTATGG CTC)和BAT-26(F:CTGCGGTAATCAAGTTTTTA G;R:AACCATTCAACATTTTTAACCC);3730XL测序分析设备(ABI 公司,美国)检测PCR 产物;Genemapper 软件分析短核苷酸重复数据。
1.3 统计学分析
使用R 软件(版本3.6.2)(https://www.r-project.org/)完成突变谱绘制,应用SnapGene、GeneMapper 软件进行突变信息分析。
2 结果
2.1 遗传性肿瘤相关基因的已知致病性或疑似致病性胚系突变
85 例符合中国人Lynch 综合征临床标准的家族性CRC 患者,经二代测序平台所检测到的3 级及以上的突变,即临床意义不明、疑似致病和已知致病突变见图1。其中,24 例(28.2%)患者携带MMR 基因已知致病性或疑似致病性胚系突变,即可确诊为Lynch 综合征,具体包括17 例MLH1相关Lynch 综合征、6 例MSH2相关Lynch 综合征以及1 例由于EPCAM基因第8~9 号外显子大片段缺失而导致的Lynch 综合征(上述Lynch 综合征患者胚系致病突变信息见表1)。

图1 85 例入组患者胚系突变情况(仅包括已知致病性、疑似致病性和临床意义不明突变)
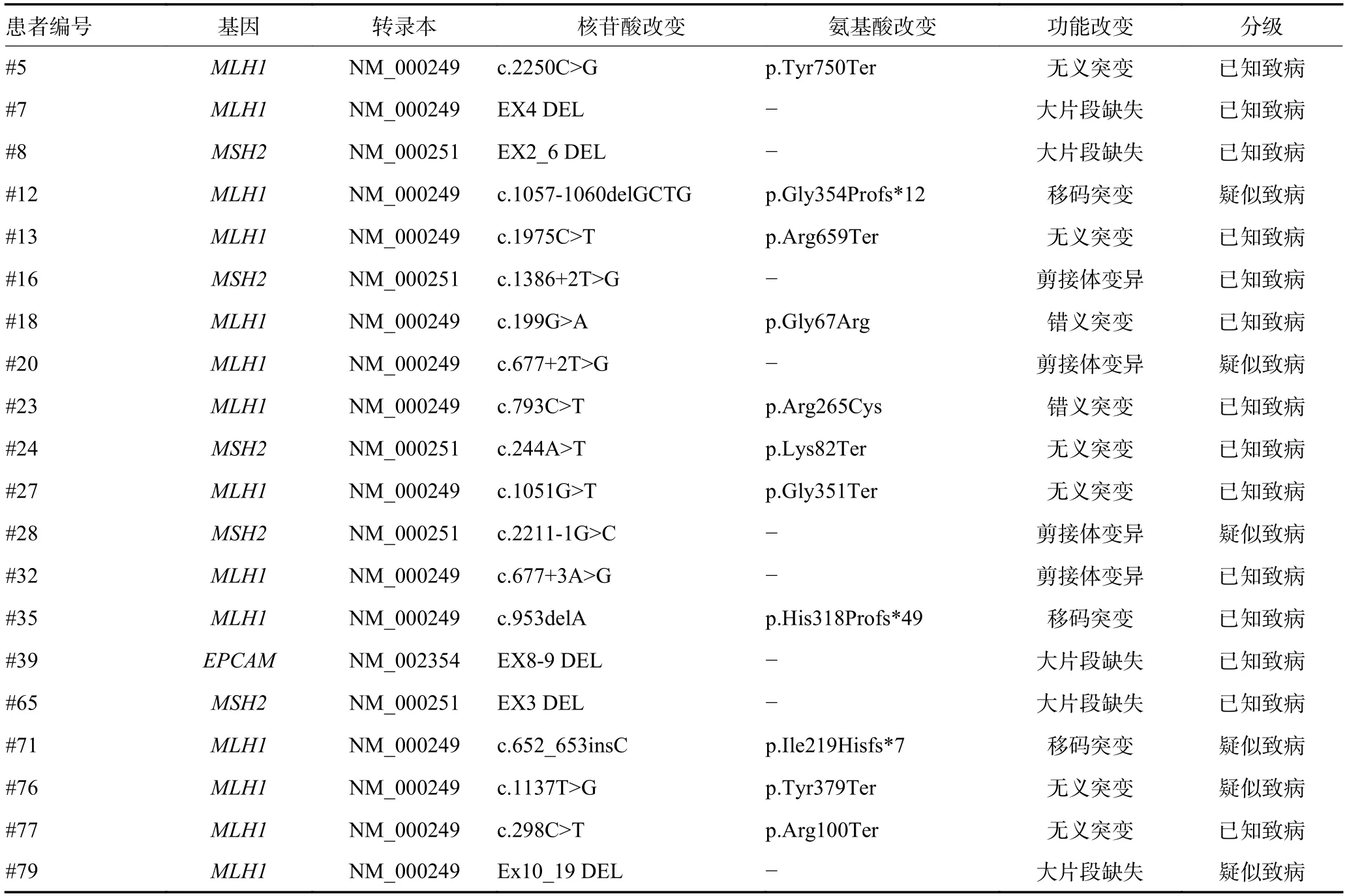
表1 携带Lynch 综合征相关基因的已知致病或疑似致病突变的CRC 患者信息

表1 携带Lynch 综合征相关基因的已知致病或疑似致病突变的CRC 患者信息 (续表1)
另外,本研究还检测到具有CRC 和肝癌家族史的第34 号直肠癌患者携带MUTYH基因c.467G>A杂合性无义突变,而未检测到其他突变。虽然MUTYH相关息肉病是一种常染色体隐性遗传病,但是该基因单等位基因突变可以增加CRC 患病风险[9]。该突变导致MUTYH基因编码蛋白在第156 位发生提前终止,造成其多肽链截短,而正常的MUTYH基因可编码549 个氨基酸。Guan 等[10]在1 例CRC 患者中检测到MUTYHp.Trp156Ter 突变,故判定该变异为已知致病突变。第76 号先证者除MLH1基因无义突变外,还携带遗传性癌症易感综合征相关基因RAD50基因[11,12]的移码突变(c.2980_2983delAAAG;p.Glu995-Argfs*2)。而第18 号先证者除MLH1基因已知致病性错义突变(c.199G>A;p.Gly76Arg)外,还携带了遗传性肾癌相关易感基因MET基因[13]的剪接体变异(NM_001 127500;c.2784+1G>A)。
2.2 Lynch 综合征相关基因临床意义不明突变
共15 例(17.6%)患者携带临床意义不明的Lynch综合征相关基因(MLH1、MSH2、MSH6和PMS2)杂合突变,具体突变信息详见表2。其中第40 号先证者携带MLH1基因c.2240_2255 delCTGATCTATACAAAGT 杂合性通读突变,该突变可造成终止密码子的改变,从而使得编码蛋白无法在正常位置终止而继续延伸翻译;与普通的移码突变不同,其并不会导致编码蛋白截短,而是蛋白C 端延长。目前暂未发现与该变异有关的功能研究和临床意义的文献报道,且在千人基因组数据库中的检出频率为0。不过,既往曾有两个关于MLH1基因通读突变的案例报道,分别报道MLH1c.2250_2251 ins AA(共编码782 个氨基酸)[14]和c.2262_2263 delGA(共编码788 个氨基酸)[15],均被判读为致病突变。为了明确MLH1基因c.2240_2255-delCTGATCTATACAAAGT 性质,本研究进行了家系验证,该家系中有多人患结直肠癌、胃癌等Lynch综合征相关恶性肿瘤,且家系中年龄最小的恶性肿瘤患者仅26 岁,符合Amsterdam Ⅱ标准;而此MLH1基因通读突变在此高度怀疑Lynch 综合征家系中存在显著的家系共分离,家系验证结果详见图2A。另外,本文采用多重荧光PCR 毛细管电泳法检测了先证者肿瘤组织微卫星状态,结果显示5 个微卫星位点中3 个位点(NR-21、NR-24 和BAT-26)不稳定,故判读为MSI-H,峰图详见图2B。因此,本研究认为此变异可能是该家族性CRC 家系的关键致病原因,但是MLH1基因编码蛋白的延长导致DNA 错配修复系统功能缺陷的具体机制有待研究者进一步的探索。

表2 携带Lynch 综合征相关基因的临床意义不明突变的CRC 患者信息
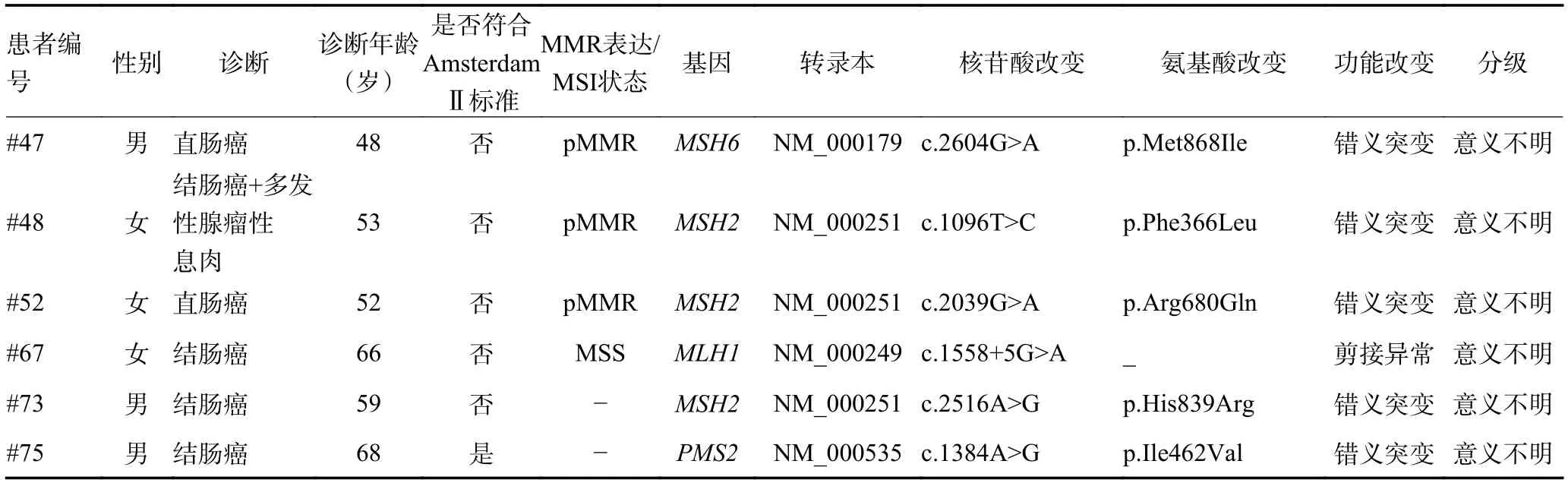
表2 携带Lynch 综合征相关基因的临床意义不明突变的CRC 患者信息 (续表2)

图2 第40 号先证者家族史和肿瘤组织微卫星状态检测结果
除第40 号家系外,其余14 个符合中国人Lynch综合征临床标准的CRC 家系均未达到Amsterdam Ⅱ标准,且MMR 蛋白表达或微卫星状态均未提示存在错配修复功能缺陷。因此,目前尚无法判断上述变异与家族性CRC 的因果关系。
2.3 其他相关基因(MLH3、MSH3)的突变
本研究中共有9 例患者被检测到携带MLH3或MSH3基因胚系突变(表3)。虽然第36 号先证者携带的是MLH3基因纯合移码突变,该变异可造成MLH3基因编码蛋白截短,但是该患者肿瘤组织呈现错配修复功能完整(proficient MMR,pMMR)且微卫星稳定(microsatellite stable,MSS),说明MLH3 蛋白对DNA 错配修复系统影响甚微。另外前期本研究组通过一代测序进行家系验证[16],发现该变异无明确家系共分离,因此,目前尚缺乏足够证据提示MLH3c.615delA 是家族性CRC 致病突变。

表3 携带MLH3 或MSH3 基因突变的CRC 患者信息
第54 号先证者同样来自一个具有显著肿瘤相关家族史的家系,家系图谱详见图3。二代测序及实时荧光定量PCR 均提示该患者携带MLH3第2~11 号外显子重复变异。然而,由于先证者的姐姐(Ⅲ-7)于43 岁时确诊卵巢子宫内膜样癌,且与先证者一样,肿瘤组织免疫组织化学检测结果显示MSH2 和MSH6蛋白表达缺失,但未携带该重复变异。因此,认为该MLH3重复变异与该家系的肿瘤家族史无直接联系。而后,对这个家系多名成员(Ⅲ-6、Ⅲ-7;父系Ⅱ-4、Ⅲ-3;母系Ⅱ-9、Ⅲ-12)的胚系基因组DNA 以及Ⅲ-7 患者肿瘤组织DNA 进行了全外显子检测。无论是父系还是母系,均未筛选出Ⅲ-6、Ⅲ-7 与父系或母系中患者所共有且健康人未携带的致病或疑似致病突变。另外,本研究发现Ⅲ-7 的肿瘤组织同时存在MSH2移码突变(NM_000251.2;c.2205_2209delCCTCA;p.I735fs)和第8 外显子拷贝数异常,不排除Ⅲ-7 肿瘤组织免疫组织化学检测的dMMR 来自MSH2基因的双等位基因体细胞突变。

图3 第54 号患者家系图谱
前文已提及的第40 号先证者,除携带MLH1基因c.2240_2 255 delCTGATCTATACAAAGT 通读突变外,同时检出MSH3基因c.2731T>G 错义突变,但是综合比较两种变异,本研究认为MLH1通读变异对DNA 错配修复系统的影响相对更大。
3 讨论
本研究共入组了85 例符合中国人Lynch 综合征临床标准的家族性CRC 患者,采用二代测序平台共确诊24 例(28.2%)Lynch 综合征,即检测到明确的致病性胚系突变。该确诊率与既往利用一代测序的研究(确诊率为23.0%~29.4%)[3,7,17]相比,显示二代测序虽能显著提升Lynch 综合征检测效率,但并不会提高其确诊率。近年来,国内也有数项利用二代测序平台进行Lynch 综合征筛查的研究[18,19],报道了中国人群的Lynch 综合征临床病理特征、突变谱等信息,但上述研究均以免疫组织化学染色提示dMMR 的CRC 患者作为筛查对象。本研究是目前国内首个以符合中国人Lynch 综合征临床标准的家族性CRC 患者作为研究对象,并采用二代测序平台描绘中国人群家族性CRC 胚系突变谱的多中心研究。
另外,15 例(17.6%)患者携带Lynch 综合征相关基因(MLH1、MSH2、MSH6、PMS2和EPCAM)临床意义不明的胚系突变,其中11 例(73.3%)突变为错义突变。如果要明确这些突变与家族性CRC 的关系,对血缘亲属样本进行家系验证是重要的一个环节。此外,MMR 基因错义突变对于DNA 错配修复系统功能的影响检测也是主要证据之一。有研究[20-21]发展一种无细胞体外MMR 活性检测技术(cell freein vitroMMR activity,CIMRA),用于评估MMR 基因突变是否影响错配修复功能,MMR 错义突变检测正确率达65%,CIMRA 技术联合数据模型后能将这一数值提高到87%。通过这项技术,Lynch 综合征相关基因意义不明的突变是否致病可得到进一步解释。
本研究中9 例患者检测出MLH3及MSH3胚系突变,然而这两个基因是否为遗传性CRC 致病基因仍存在较大争议。MLH3 蛋白是DNA 错配修复基因的MutL 同源(MutL homolog,MLH)家族的成员之一,而MSH3 蛋白与MSH2 异源二聚化以形成MutSβ 二聚体,参与DNA 错配修复[22-23]。但是研究显示MLH3和MSH3 蛋白对于DNA 错配修复的贡献非常有限,多个可能与遗传性CRC 相关的MLH3基因突变被认为既不影响蛋白表达水平,也不影响其功能[24,25]。一项研究称MLH3c.3563C>G p.Ser1188Ter 可能是遗传性息肉病的致病原因[26]。然而本研究中的第36 号和第54 号患者分别携带MLH3c.615delA 纯合突变和第2~11 外显子重复变异,通过家系验证等变异分析手段,认为尚缺乏充足证据证明MLH3基因在遗传性CRC 中的致病意义。
此外,二代测序的高速发展除可以实现利用较少的生物样本以检测更多的基因以外,也会带来大量临床意义不明的突变;还有一部分患者和(或)其血缘亲属的症状是某些遗传性肿瘤综合征的典型临床表型,但通过常规的基因检测手段未能发现相关基因的突变。以上两个现象均会给临床医师和患者及其亲属带来巨大的困惑,如何去甄别、判读、寻找真正的发病原因是一个亟待解决的难题。首先,强烈建议尽可能多的血缘亲属提供样本和临床信息,尤其是患肿瘤的血缘亲属和年龄较大的未患恶性肿瘤的血缘亲属形成对照,协助判断突变是否存在家系共分离。其次,面对临床表型典型但致病突变未明的患者,需要考虑到检测方法的局限性,可以选择检测范围更广的手段,如全外显子测序或全基因组测序等方法,除寻找其他基因的致病变异以外,同时可以检测已知关键基因是否发生拷贝数变异、5’UTR、启动子区和3’UTR 等一些容易被遗漏的突变。
本研究通过二代测序平台检测符合中国人Lynch综合征临床标准的家族性CRC 患者中最终确诊为Lynch 综合征的比例,确诊率与既往一致,但同时发现存在较多Lynch 综合征相关基因意义不明的突变;家系验证和肿瘤组织DNA 错配修复系统检测(MMR 蛋白表达水平或微卫星状态)是目前最有效的方法,可以帮助判断基因突变是否与疾病直接相关。另外,需要谨慎对待尚未明确的基因突变检测结果,并认识到所用检测方法的局限性,必要时进行更深入的家系研究和更全面的分子检测。
