富锂锰基正极材料反应机理研究进展
2022-12-22杨凤玉陈勃涛
杨凤玉,董 华,陈勃涛
(1.天津国安盟固利新材料科技股份有限公司,天津 301811;2.天津市固态电池关键材料与技术企业重点实验室;3.天津荣盛盟固利新能源科技有限公司)
2021 年全国两会上“碳达峰”、“碳中和”的“双碳”目标首次被写入政府工作报告。2021年被称为“碳中和”元年,碳中和战略已经奠定了中国新能源行业的发展基调,这为锂离子电池的发展带来了新的机遇和更广阔的市场。目前,锂电池主要正极材料中钴酸锂(LCO)、三元材料(NCM)最高比容量只能达到220 mA·h/g 左右,提升空间有限,而且材料的成本高,考虑到资源的有限性,降成本空间小。磷酸铁锂(LFP)和锰酸锂(LMO)虽然成本优势明显,但是比容量较低,电池能量密度提升较为困难,无法满足未来能量密度越来越高的要求。高能量密度、低成本、高安全性仍将是锂离子电池未来发展的主流方向[1-2]。
富锂锰基正极材料[xLi2MnO3·(1-x)LiMO2]具有较高的比容量(≥250 mA·h/g),是实现电池能量密度达到500 W·h/kg 目标的一个重要选择。而且该材料以Mn 元素为主,Ni 和Co 等稀有元素的含量可以大幅度降低,最大程度地降低了材料的成本。富锂锰基正极材料由于具有高能量密度、低成本与资源的可持续性,可能成为下一代锂离子电池的主流正极材料[3-4]。虽然富锂锰基正极材料表现出良好的应用前景[5-6],但是其存在的问题也较为突出。该材料首次不可逆容量高、倍率性能较差、循环过程中放电中值电压不断衰减[7-8]等问题仍然十分严重,电压迟滞导致能量转换效率低[9]、全电池体系产气严重、高电压下电解液分解等问题严重阻碍了其量产应用。为了应对这些挑战,了解材料的反应机理及脱嵌锂机制变得至关重要。然而,对富锂锰基正极材料的相关综述大多是关于材料的合成与改性[10-11],与反应机理相关的综述[12]较少,对具体的电化学反应过程的相关阐述缺少系统化的总结。因此,笔者对富锂锰基材料的结构、反应机理两个方面进行了总结归纳,并且对富锂锰基材料未来的产品化应用方向提出了展望。
1 富锂锰基正极材料的结构
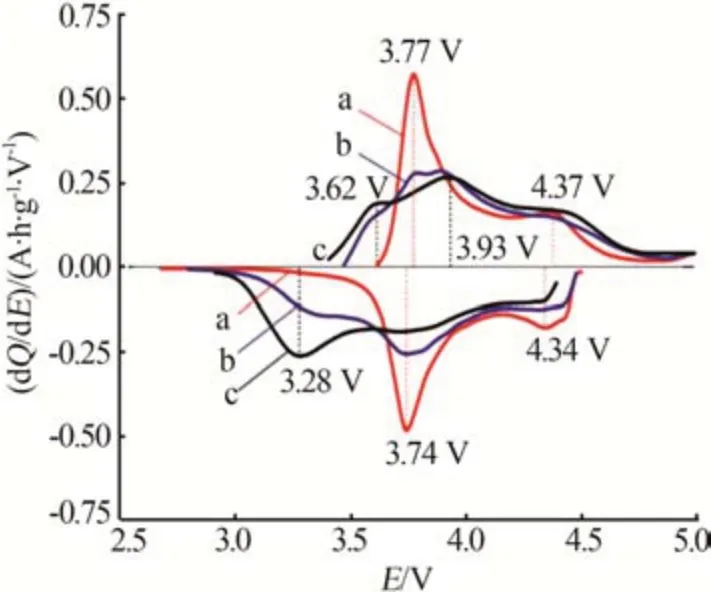
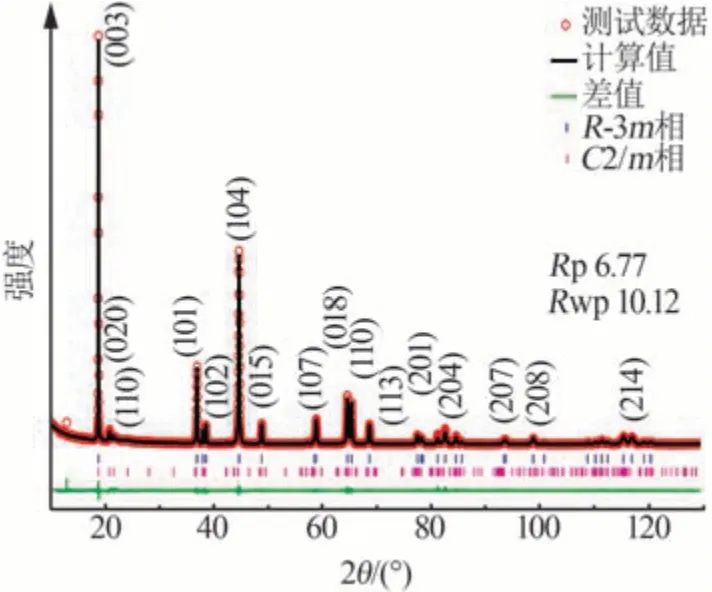
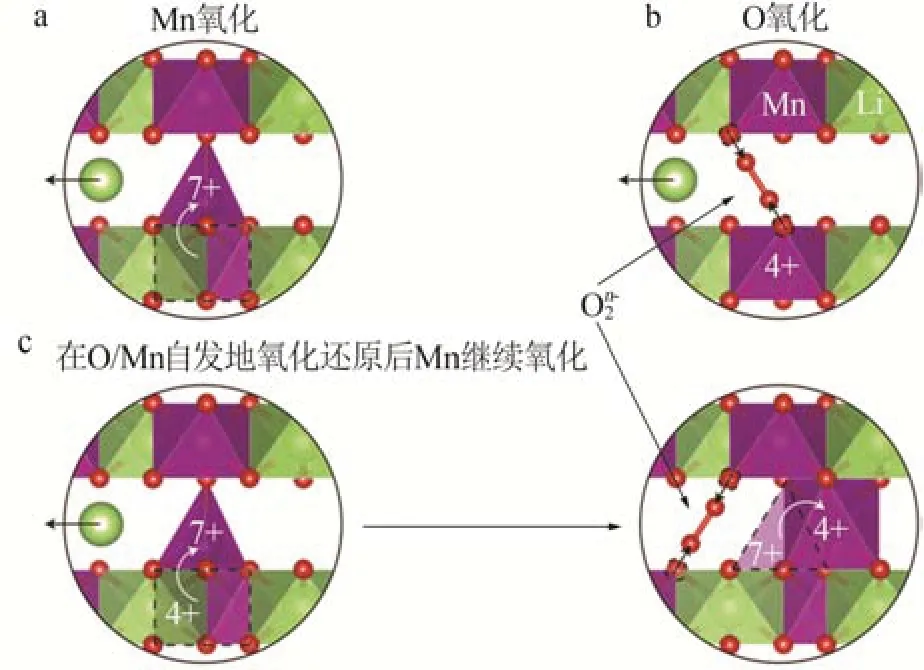
富锂锰基正极材料通常用xLi2MnO3·(1-x)LiMO2(0 2001 年,LU 等[14]发现Li1+xNi1/3-2x/3Mn2/3-x/3O2样品的a、c等晶格常数与元素比例呈线性关系,服从Vegard 定律,表明该材料具有固溶体结构特征。JARVIS 等[15]采用像差校正扫描透射电子显微镜(AC-STEM)对富锂锰基正极材料的原子排列进行观察,提供了Li[Li0.2Ni0.2Mn0.6]O2是固溶体的证据,见图1。图1显示出清晰的单相结构,堆垛层错高度集 中。2014 年GENEVOIS 等[16]也 通 过HAADFSTEM等技术手段观察到均匀的类Li2MnO3结构,Li+和Ni2+位于过渡金属(TM)层中的Li 位点,揭示了LMRO的单相固溶体特征。 图1 Li[Li0.2Ni0.2Mn0.6]O2的AC-STEM照片[15]Fig.1 AC-STEM image of Li[Li0.2Ni0.2Mn0.6]O2[15] 2004 年,KIM 等[17]通 过 固 态 核 磁 共 振(ss-NMR)测试表明xLi2TiO3·(1-x)LiNi0.5Mn0.5O2样品中存在Li2TiO3畴,这为两相纳米畴的概念提供了依据,Li2MnO3和NCM结构的选区电子衍射(SAED)图案差异很大。2014 年YU 等[18]通过HAADF-STEM也证实了Li1.2Ni0.15Co0.1Mn0.55O2中存在两相结构,见图2。由图2 可以观察到一些区域具有连续的点对比度(区域Ⅰ和Ⅲ),而其他一些区域具有不连续的点对比度(区域Ⅱ和Ⅳ),Li1/3(Mn)2/3板反映在不连续区域中,而LiMO2组分中的M(过渡金属离子)反映在连续区域中,这清楚地揭示了平面内类Li2MnO3和类LiMO2区域的共存。 图2 Li1.2Ni0.15Co0.1Mn0.55O2的HAADF-STEM照片[18]Fig.2 HAADF-STEM image of Li1.2Ni0.15Co0.1Mn0.55O2[18] 关于层状富锂锰基材料的结构是固溶体还是两相的争论一直没有停止。GREY 小组通过ss-NMR技术对Li[Li(1-2x)/3NixMn(2-x)/3]O2进行了测试,结果表明在化学位移为1.560×10-3处有更强的共振峰,表明TM层中的Li+更倾向于被Mn4+而非Ni2+包围,进一步 证 明 了Li[Li(1-2x)/3NixMn(2-x)/3]O2电 极 中 存 在Li(OMn)6短程有序的结构[19]。2014 年MOHANTY等[20]采用中子衍射技术进一步证实M 层存在有序排列的阳离子。 2013年,MCCALLA等[21]绘制了Li-Mn-Ni-O体系某些实验条件下的相图,以相图为指导根据合成条件选择层状区域边界附近的组合物是层状纳米复合材料或是单相层状。除元素比例外,材料的结构还与制备条件等其他参数密切相关,尤其是与合成方法、退火温度[22]、过渡金属类型、化学计量学材料尺寸等相关因素密切相关,不能抛开材料的体系与合成条件去单纯讨论材料的结构[23-24]。 富锂材料Li2MnO3与传统的锂层状氧化物相比,过量的Li+会导致过渡金属离子的高价态,因此这些材料最初并未被设想为锂离子电池的潜在正极。THACKERAY 团队于1991 年尝试通过酸处理从Li2MnO3中 浸 出Li2O 来 获 取LiMnO2,发 现 最 终 产品Li1.09Mn0.91O2具有电化学活性,其比容量约为200 mA·h/g,使人们看到了富锂锰基材料的希 望[25]。2001 年DAHN 团 队 首 先 将 一 系 列L[iNixLi(1/3-2x/3)Mn(2/3-x/3)]O2电极在第一次循环中充电至4.8 V,并发现电压为2.0~4.6 V、55 ℃时具有220 mA·h/g 的高可逆比容量[26]。从此富锂锰基材料的高比容量属性走入人们的视野,也引发了广大研究者的兴趣。2004 年THACKERAY 提出富锂层状固溶体正极材料这个概念,系统介绍了xLi2M′O·3(1-x)LiMn0.5Ni0.5O2电 极(M′=Ti,Mn,Zr,0≤x≤0.3)的结构和电化学性能,锂电池循环时xLi2M′O·3(1-x)LiMn0.5Ni0.5O2(x=0.3)电极的比容量在4.60~1.45 V超过300 mA·h/g[17]。 层状富锂锰基正极材料普遍能够实现大于200 mA·h/g 的放电比容量,其中一些甚至能够达到400 mA·h/g 的超高比容量[27],这种超高比容量无法用常规的阳离子脱嵌机理解释,而且材料充放电的微分曲线显示出明显不对称的氧化还原峰(见图3)[28],存在富锂锰基材料特有的反应机理。目前对富锂锰基材料的反应机理主要有Li+/H+交换理论、阴离子氧氧化还原机理(阴离子电荷补偿理论)[29-30]和Mn4+/Mn7+反应机理假说。 图3 不同正极材料第2次循环的微分曲线[28]Fig.3 Differential curve of different cathode materials at second cycle[28] 2002年,ROBERTSON等[31]研究了Li2MnO3在非水介质中的电化学活性,对充电脱出Li+和放电结束时的锂锰氧化物电极进行了X 射线光电子能谱(XPS)分析。结果表明,锰在整个过程中保持+4价,在充电后期测定出氢(H)元素的存在,其含量与脱出的Li 量以及容量值有很强的线性对应关系,因此认为该反应机理既不涉及Mn4+—Mn5+的氧化反应,也不涉及O2-的同时去除,而是通过H+交换Li+,H+在电解液中产生。2007年,层状富锂锰基氧化物[L(iLixNiyCozMn1-xyz)O2,LMROs]在高电压范围(4.5~4.8 V)工作时,Li+—H+交换机制被证明是一种寄生副反应,同时这种质子交换机理也对高温环境下富锂锰基材料优越的电性能进行了较好的解释[4]。 基于在高电位LixCoO2中观察到O—O距离略微缩短,氧的氧化还原被提出[32]。富锂锰基正极材料超高的放电比容量难以用常规的阳离子氧化还原完全证实,阴离子参与电化学反应的设想也陆续被提出。2009年,KOYAMA等[33]首次提出了富锂锰基材料的阴离子晶格氧参与电荷补偿过程的观点,通过密度泛函理论(DFT)计算Li2-xMnO3(x=2.0、1.5、1.0、0.5)的电子结构,提出Mn4+保持稳定、晶格氧的氧化有助于Li+脱出过程中的电荷补偿。2013 年,TARASCON 课题组将Li2Ru1-ySnyO3作为模型材料实验性地揭示了LMRO 中存在晶格氧氧化还原反应,Li2Ru1-ySnyO3电极在不同充放电状态下的XPS显示,在充电至4.6 V 的曲线中出现一个位于530.5 eV 的新峰(见图4a、b),鉴定为过氧/超氧类物质,起源于晶格氧的氧化[34]。然而,具体的反应机制一直存在争议[35-36]。 图4 Li2Ru1-ySnyO3电极在不同充放电状态下的XPS图(a、b);充电过程中Li2Ru0.5Sn0.5O3中产生类过氧化物示意图(c、d)[34]Fig.4 (a、b)XPS spectra of Li2Ru1-ySnyO3electrode under different charge and discharge conditions,(c、d)scheme of generation of peroxides⁃like in Li2Ru0.5Sn0.5O3 during charging process[34] 2.2.1 晶格氧析出理论与多步反应机制 一般的富锂锰基正极材料首次充电会出现两段平台:第一段平台电压小于4.5 V,此段平台呈上升趋势,Li+从材料锂层中脱出,同时伴随着过渡金属的氧化还原;第二段平台电压为4.5 V 左右,这一现象在随后的充放电循环中消失。2002年,LU等[37-38]通过电化学研究、粉末中子衍射(ND)和原位X射线衍射(XRD)等技术手段,对电极材料的初始结构、电化学和结构演化进行了综合分析,解释了初始充电过程中的不可逆平台(即“激活平台”)和额外容量,提出了晶格氧析出理论,这对LMRO 的后续研究具有深远的影响。2006年BRUCE课题组通过原位差分电化学质谱法(DEMS)发现L[iNi0.2Li0.2Mn0.6MnO2]材料在活化过程中析出了O2,再一次证明了以上观点[39]。 2007 年THACKERAY 等[40]系统整理了富锂锰基正极材料的多步反应机制,该理论依赖于Li2MnO3和LiMO2组成的相图。对于典型的富锂锰基正极材料[xLi2MnO3·(1-x)LiMO2],在第一次充电过程中在4.4 V之前锂离子从LiMO2相中连续脱出,在4.4 V之后出现了一个独特且可区分的电压平台(见图5),代表由复杂的化学反应和结构演变引起的氧释放和锂离子从Li2MnO3相中脱嵌,高电压充电导致Li2O脱嵌、产生氧元素缺陷,从而得到活化的MO2,显示出高容量。图5 中的放电过程表明,锂离子重新插入MO2结构中,在循环中锂离子的可逆插入/提取显示出较小的电压滞后以及在4.5 V处没有电压平台,表明Li2MnO3活化后的不可逆性。 图5 xLi2MnO3·(1-x)LiMO2电极电化学反应路径的组成相图[40]Fig.5 Compositional phase diagram of electrochemical reaction pathways for xLi2MnO3·(1-x)LiMO2electrode[40] 关于Li2MnO3的可逆性存在另外一些观点。MUHAMMAD等[29]从原子水平研究了高容量富锂锰基氧化物0.4Li2MnO3·0.6LiMn0.5Ni0.5O2的反应机理,图6 为0.4Li2MnO3·0.6LiMn0.5Ni0.5O2的测试与拟合精修XRD谱图。由图6看出,样品XRD谱图中在2θ为20~30°的衍射峰通常归因于TM 层中的超晶格结构。大多数情况下,样品XRD谱图中的超晶格峰在平台结束时消失,但是在Li1.2Ni0.2Mn0.6O2电极的同步加速器XRD 谱图中可以很好地观察到它们在整个充电/放电过程中保持良好。2009 年,YU 等[41]通过研究Li2MnO3在不同充电状态下的初始放电比容量,提出Li+是同时从Li 层和TM 层脱出的,并且在放电过程中没有Li+重新进入TM 层。因此,关于过量Li+从TM层中脱出时所需的相对电位以及这种脱出是否可逆等问题的相关争论仍然存在。 图6 0.4Li2MnO3·0.6LiMn0.5Ni0.5O2的测试与拟合精修XRD谱图[29]Fig.6Testing and fitting refined XRD patternsof 0.4Li2MnO3·0.6LiMn0.5Ni0.5O2[29] 2.2.2 Li-O-Li 构型孤对电子理论 较多观点认为阴离子氧参与反应的方式是与金属离子杂化成键。2016年,CEDER课题组首次提出未杂化氧参与电化学反应过程,使富锂锰基正极材料获得额外容量[42]。金属层氧活性源于非常特殊的Li-O-Li 构型,见图7。由图7 可知,这种构型可以使氧产生孤对电子,证明富锂材料中的晶格氧的氧化还原是Li-O-Li构型中未杂化O 2p态中提供不稳定电子,与杂化TM-O 态无关。在锂嵌入正极中产生未杂化(孤立)氧,是开发更高能量密度正极材料的一个令人兴奋的新方向。 图7 以Li-O-Li 构型存在的活性氧参与氧化过程的结构和化学起源[42]Fig.7 Structural and chemical origin of reactive oxygen species involved in oxidation process in Li-O-Li configuration[42] 2.2.3 晶格氧还原产物为二聚体 2013 年,TARASCON 团队研究了Li2Ru1-ySnyO2(LRO)中的电荷补偿机制,通过实验揭示了LRO 中的可逆氧氧化还原反应[34]。2015年,他们利用原位电子顺磁共振(EPR)技术研究了Li2Ru0.75Sn0.25O2中氧的氧化还原反应,证明可逆O2n-物质的形成[43]。O—O 二聚体(缩短的O—O 键)首先通过带电的Li2IrO3电极中的环形明场STEM(ABF-STEM)可视化,观察到来自氧化O—O 二聚体的晶格氧键长约为0.25 nm,比Li2O2(0.15 nm)长1.6 倍,因此在这项工作中其被命名为“类过氧”物质。 ASSAT 等[44-45]认为可逆的氧基氧化还原活性在高压充电阶段起主导作用。如图8a(LMRO 材料第二次充放电微分曲线及动力学控制步骤分析)所示,氧化On-(n<2)出现在4.46 V 以上,并且在放电过程中(2.0 V)逐渐下降,至少可以提供109 mA·h/g的放电比容量。进一步循环发现氧化晶格氧的再生和消失,表明了持续和可逆的阴离子氧化还原反应性。此外,恒电流间歇滴定技术(GITT,图8b)表明,阳离子氧化还原在动力学上比阴离子氧化还原快。因此,大量阴离子活性的存在解释了LMRO正极材料的缓慢动力学和电压衰减问题。阳离子/阴离子氧化还原之间的相互作用为LMRO 正极材料高比容量背后的反应机制提供了更具体的解释。 图8 LMRO材料第二次充放电微分曲线及动力学控制步骤分析(a)、三电极体系GITT 测试(b)[44]Fig.8 Charge⁃discharge differential curve and kinetic control step analysis of LMRO material in second cycle(a);GITT test of three electrode system(b)[44] 2.2.4 晶格氧还原产物为氧空穴理论 其他研究人员认为,晶格氧化的产物不是真正的O2 2-(O—O 二聚体)类物,而是O-/On-类带有电子/空穴的氧离子。2016年,BRUCE团队结合X射线吸收光谱(XAS)和同位素电化学质谱(DEMS),确定在带电的Li1.2Ni0.13Co0.13Mn0.54O2电极中形成了由Li+/Mn4+配位的O原子上的电子/空穴态[46]。此外,在其拉曼光谱中没有O—O 的过氧振动,表明在LMRO中没有形成真正的O22-物种。GENT 等[47]研究了Li1.17-xNi0.21Co0.08Mn0.54O2富锂锰基材料,通过扫描透射X射线显微镜(STXM)、XAS以及共振非弹性X射线散射(mRIXS)等表征结果表明,电压滞后等现象源于阴离子氧化还原和阳离子迁移之间的强耦合,提出晶格氧氧化还原反应不是刚性O2-/O-氧化还原对,更可能是[O2-+TM]→[O-+TMmig]+e(-TMmig 指迁移的过渡金属元素)。 一些研究人员认为实际氧原子氧化还原的产物是O-/On-或O22-/O2n-(O—O 二聚体)是有条件的。2016 年CEDER 课题组的研究成果表明,当氧与较少量的金属离子结合时,电子/空穴会聚结形成过氧物种[46],氧空穴参与氧化还原反应,见图9。同年ISLAM 课题组的研究结果表明,不稳定局部氧空穴在脱锂开始时形成,然后锂不断脱出,氧空穴转化为过氧或超氧化物物种以及进一步的O2气体[48]。因此,他们提出防止氧二聚化是稳定O-物种和提高阴离子氧化还原反应可逆性的关键因素。 图9 Li1.2Ni0.13Co0.13Mn0.54O2材料中O-/O2-氧化还原对示意图[46]Fig.9 Schematic diagram of O-/O2-redox pair in Li1.2Ni0.13Co0.13Mn0.54O2 materials[46] 晶格氧氧化还原反应的关键是晶格氧的氧化程度和所得氧化物质的性质。在带电的3d、4d、5d 基LRO 中观察到O—O 键的缩短,并且这些键中的大多数比Li2O2中的过氧O—O键长。LRO系统的复杂性例如结构差异(层和阳离子无序)、TM 种类、化学计量比、粒径、氧氧化还原反应的深度(表面和本体)等不同,具体的反应机理及产物也会有很大的差别。关于各种富锂材料中的氧化还原反应以及电子空穴(O-/On-)和O—O二聚体(O22-/O2 n-)之间的边界,随着先进表征技术的快速发展,上述问题很可能会在不久的将来得到详细的解释。 尽管晶格氧氧化还原反应解释了富锂锰基正极材料的高容量,但是对具体的反应过程还缺少一致的认识,这引发了大家对4d 和5d 过渡金属氧化物替代反应机制的探索,因此新的Mn4+/Mn7+反应机理假说被提出来,即Mn 的氧化超过+4 价可能涉及Mn 离子在八面体和四面体位点之间的迁移,这为解释这种高比容量提供了另一种可能的机制。RADIN 等[49]提出了Mn4+/Mn7+混合相的可能形成机制,见图10。Mn4+氧化为Mn7+经历了一系列相变,其中包含Mn4+化合物(MnO2和Li4Mn5O12)和Mn7+化 合 物(Mn2O7和LiMnO4)。混 合Mn4+/Mn7+相 即Li1/2MnO3理论上可能来自Li2MnO3相,因为它只需要Mn从八面体位点到四面体位点的单一迁移。 图10 LMRO材料中的Mn4+/Mn7+反应机制假说[49]Fig.10 Hypothesis of Mn4+/Mn7+reaction mechanism in LMRO materials[49] 基于未来更加先进的光谱、成像技术和相关检测技术的发展,富锂锰基阴离子氧化还原的一些基础问题需要更多地被关注与研究,量化活性氧与缺陷之间的联系,进一步抑制缺陷的产生、探究活性氧对容量的贡献以及缺陷如何平衡等一系列问题的解决,将对富锂锰基材料的实用化进展具有重大意义。 富锂锰基正极材料xLi2MnO3·(1-x)LiMO2(M=Ni,Co 或Mn)以其高比容量的潜力成为锂离子电池新一代正极材料的有力竞争者。近年来的研究已经在材料合成、掺杂和包覆改性方面取得了很大进步[50-52],氧化的晶格氧、氧空位、过渡金属迁移、层状到尖晶石的转变、“两相机制”和晶格演化等具体的反应机理还需要进一步研究与探讨。优化富锂材料的结构稳定性,抑制O2释放、过渡金属迁移[53]、分层到尖晶石相转变也是富锂材料产业化过程较大的技术障碍。 由于不同合成条件和化学计量会造成材料局部结构的差异与转变[54],结构的差异及转变机制对电性能的表现和电荷补偿仍然难以捉摸[55-57]。对工业化生产提出了新的挑战,前驱体的批次稳定性、理化指标的公差范围要比常规三元材料的要求更为严格。材料的合成过程中各个参数需要进行精确控制,对生产过程的管控和过程能力的要求更高,对成品检测也要求更加深入的表征手段。 富锂锰基材料独特的反应机理与常规的正极材料差别较大,适合的化成活化制度与后续电池的容量发挥及循环稳定性息息相关。反应过程中涉及到活性氧参与到反应过程中,在负极的挑选过程中较为稳定的碳负极、无“催化活性”对体系的产气也尤为重要。电解液与富锂材料的相容性以及是否存在诱导性,对全电的设计提出了更高要求。富锂锰基正极材料以其独特的组成和结构具有巨大的应用潜力,但是在材料的反应机理、结构控制、产业化应用等方向仍然需要踏实深入地研究,高性价比的富锂锰基材料的产业化应用将再一次推动锂离子电池发展迈向一个新台阶。1.1 单相固溶体观点

1.2 两相混合物观点

1.3 局部阳离子有序排列
1.4 结构与元素比例和制备条件等密切相关
2 富锂锰基材料的反应机理
2.1 Li+/H+质子交换理论
2.2 阴离子氧氧化还原机理





2.3 Mn4+/Mn7+反应机理假说
3 结语与展望
