Small-molecule 7,8-dihydroxyflavone counteracts compensated and decompensated cardiac hypertrophy via AMPK activation
2022-12-20PengZhouHANGPeiFengLIJieLIUFengFengLITingTingCHENYangPANManRuZHANGHuaQingYUHongYuJIZhiMinDUJingZHAO
Peng-Zhou HANG, Pei-Feng LI, Jie LIU, Feng-Feng LI, Ting-Ting CHEN, Yang PAN,Man-Ru ZHANG, Hua-Qing YU, Hong-Yu JI, Zhi-Min DU,4,, Jing ZHAO,5,
1. Institute of Clinical Pharmacology, the Second Affiliated Hospital of Harbin Medical University (University Key Laboratory of Drug Research, Heilongjiang Province), Harbin, China; 2. Department of Pharmacy, Clinical Medical College,Yangzhou University, Northern Jiangsu People's Hospital, Yangzhou, China; 3. Department of Pharmacology, Harbin Medical University, Harbin, China; 4. State Key Laboratory of Quality Research in Chinese Medicines, Macau University of Science and Technology, Macau, China; 5. Department of Cardiology, the First Affiliated Hospital of Harbin Medical University, Harbin, China
ABSTRACT BACKGROUND Pathological cardiac hypertrophy is a compensated response to various stimuli and is considered a key risk factor for heart failure. 7,8-Dihydroxyflavone (7,8-DHF) is a flavonoid derivative that acts as a small-molecule brain-derived neurotrophic factor mimetic. The present study aimed to explore the potential role of 7,8-DHF in cardiac hypertrophy. METHODS Kunming mice and H9c2 cells were exposed to transverse aortic constriction or isoproterenol (ISO) with or without 7,8-DHF, respectively. F-actin staining was performed to calculate the cell area. Transcriptional levels of hypertrophic markers, including ANP, BNP, and β-MHC, were detected. Echocardiography, hematoxylin-eosin staining, and transmission electron microscopy were used to examine the cardiac function, histology, and ultrastructure of ventricles. Protein levels of mitochondria-related factors, such as adenosine monophosphate-activated protein kinase (AMPK), and peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), were detected. RESULTS 7,8-DHF inhibited compensated and decompensated cardiac hypertrophy, diminished the cross-sectional area, and alleviated the mitochondrial disorders of cardiomyocytes. Meanwhile, 7,8-DHF reduced the cell size and repressed the mRNA levels of the hypertrophic markers of ISO-treated cardiomyocytes. In addition, 7,8-DHF activated AMPK and PGC-1α signals without affecting the protein levels of mitochondrial dynamics-related molecules. The effects of 7,8-DHF were eliminanted by Compound C, an AMPK inhibitor. CONCLUSIONS These findings suggest that 7,8-DHF inhibited cardiac hypertrophy and mitochondrial dysfunction by activating AMPK signaling, providing a potential agent for the treatment of pathological cardiac hypertrophy.
Cardiac hypertrophy is an adaptive response to the excessive cardiac hemodynamic load and maintenance of cardiac function.[1]The myocardium initially undergoes hypertrophic growth as a compensatory response to improve myocardial contractility, reduce wall stress and maintain cardiac output. By contrast, sustained pathological stimulation can increase oxygen consumption, damage energy metabolism, and eventually lead to decreased cardiac function and heart failure.[2]The inhibition of the transition from cardiac hypertrophy to heart failure by regulating mitochondria and oxidative stress is currently applied to improve the quality of life and reduce the mortality rate of patients.[3]Accumulating studies have revealed that dysfunctional mitochondrial bioenergetics is one of the primary determinants of pathological cardiac hypertrophy.[4]Cardiac hypertrophy disrupts the relationship between ATP consumption and production, and mitochondrial bioenergy must keep pace with the phenotype of cardiac hypertrophy.[5]The adenosine monophosphate-activated protein kinase (AMPK) is a central regulator in governing the balance of ATP consumption and production of multiple metabolic pathways in cardiac hypertrophy.[6,7]AMPK inhibits cardiac hypertrophy and restores energy balance by repressing various anabolic signals, including the mammalian target of rapamycin (mTOR)/p70 ribosomal S6 protein kinase (p70S6K) and eukaryotic elongation factor-2 (eEF2), as well as activating catabolic pathways, such as glycolysis.[7,8]AMPKα2 inhibits the progression of heart failure by promoting mitophagy via Ser495 phosphorylation in PTEN-induced kinase 1 (PINK1).[9]Notably, cross-talk between AMPK and reactive oxygen species (ROS)in heart injury is well-documented. ROS activates AMPK, and AMPK inhibits excessive ROS production.[10-12]
Numerous studies have verified the crucial role of brain-derived neurotrophic factor (BDNF) in both cardiac physiology[13,14]and diseases.[15,16]Nonetheless, whether BDNF plays a primary role against cardiac hypertrophy is not well defined. The flavonoid derivative, 7,8-dihydroxyflavone (7,8-DHF) is a natural compound first reported as a mimetic of BDNF.[17]Its beneficial role in various neurodegenerative disorders such as Parkinson’s disease and Alzheimer’s disease has been extensively explored.[18]In previous studies, we found that 7,8-DHF protected against doxorubicin-induced cardiotoxicity and myocardial ischemic injury by enhancing mitochondrial oxidative phosphorylation or alleviating excessive mitochondrial fission.[19,20]
On the basis of the aforementioned findings, the effect of 7,8-DHF on compensated and decompensated pathological cardiac hypertrophy is relevant and should be studied to reveal the underlying mechanisms. We hypothesize that 7,8-DHF inhibits pathological cardiac hypertrophy by regulating AMPK-dependent mitochondrial function.
METHODS
Reagents
7,8-DHF (purity 99.9%) was purchased from MedChemExpress (HY-W013372, Shanghai, China).Isoproterenol (ISO) was provided by Aladdin Biochemical Technology Co., Ltd (#129810, Shanghai,China). Compound C, an AMPK inhibitor, was supplied by MedChemExpress (HY-13418A, Shanghai,China). The antibody against the atrial natriuretic peptide (ANP) was purchased from BOSTER Biological Technology (#A01318-1). Antibodies for p-AMPKα (Thr172, #2535), AMPKα (#2532), p-signal transducer and activator of transcription 3 (STAT3,Tyr705, #9131), STAT3 (#4904), mitofusin 2 (Mfn2,#9482), dynamin-related protein 1 (Drp1, #5391),and cytochrome c oxidase subunit IV (COX-IV, #4850)were supplied by Cell Signaling Technology (Danvers,MA, USA). The followings were also purchased: the peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) antibody from Abcam (ab54481,Cambridge, USA); the antibody against OPA1 from BD Transduction Laboratories (#612606, Lexington,KY, USA); the antibody against mitochondrial fission 1 protein (Fis-1, #DF12005) from Affinity Biosciences; and the anti-GAPDH (TA-08) antibody from ZSGB Co. Ltd (Beijing, China). Phalloidin-iFluor 488 was provided by Abcam (ab176753, Cambridge, USA). Wheat germ agglutinin (WGA) was supplied by Sigma (L4895, USA).
Transverse Aortic Constriction Mouse Model
All animal experiments were performed following the standard protocols approved by the Animal Care and Use Committee of the Second Affiliated Hospital of Harbin Medical University (No. KY2018-067). Mice were raised according to the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health (revised in 2011). A transverse aortic constriction (TAC)-induced cardiac hypertrophy model was established in Kunming mice (aged 8 weeks, male) using a previously reported method.[21]All mice were reared under standard conditions and adapted to the environment for 1 week. After being anesthetized with 1.2% avertin (2, 2, 2-tribromoethanol, Sigma), the mice were fixed on the operating plate in the supine position, and the tissue and muscle around the thoracic aorta were bluntly dissected under sterilizing conditions. After the mice underwent midline sternotomy, a 27-gauge needle was tied to the aorta between the innominate and left common carotid arteries by using a 6-0 silk suture. The cushion needle was drawn out, and the sternum and skin were immediately sutured with the ligation thread. The procedures conducted in the sham group were the same, except for ligation. Two batches of TAC experiments were performed. Three days after TAC in each batch of experiments, mice were randomly divided into two groups for continuous observation for 4 or 8 weeks. Thus, in the follow-up experiments,the detailed animal groups were included in 1 batch:the sham, TAC, and +7,8-DHF (5 mg/kg, i.p., daily for 8 weeks after TAC) groups (n= 6 per group); the other batch: sham, TAC, and +7,8-DHF (5 mg/kg,i.p. daily for 4 weeks after TAC) groups (n= 10 in the sham group,n= 13 in the TAC and +7,8-DHF groups). At the end of the observation, echocardiography was conducted, and murine hearts were collected immediately and then stored in stationary fluid or a refrigerator at -80 °C for subsequent experiments.
Echocardiography
The left ventricular function was examined using an echocardiographic system with an ultrasound machine Vevo2100 (VisualSonics, Toronto, Canada)equipped with a 10 MHz phased-array transducer with the M-mode recordings as previously described.[22]The average of at least three consecutive cardiac cycles was used for all ultrasound measurements. Echocardiographic parameters included the end-diastolic left ventricular volume (LVV,d), endsystolic left ventricular volume (LVV,s), left ventricular end-diastolic posterior wall thickness (LVPW,d),left ventricular end-systolic posterior wall thickness(LVPW,s), end-diastolic left ventricular internal diameter (LVID,d), and end-systolic left ventricular internal diameter (LVID,s). Ejection fraction (EF) was measured automatically by the machine, and fractional shortening (FS) was calculated using the following equation: ((LVID,d - LVID,s)/LVID,d) × 100.
Morphological and Histological Analysis
The heart weight (HW) and tibia length (TL) was measured, and TL is a reference for the ages of the mice. The HW-to-TL ratio (HW/TL) was calculated.After being washed with a saline solution, the isolated hearts were fixed in 10% formalin, embedded in paraffin, and then sectioned transversely at a thickness of 5 μm. Hematoxylin-eosin (HE) staining was then performed to observe the pathological changes in the groups. Images were captured under an Olympus BX53 fluorescence microscope (Japan) equipped with a DP80 camera.
Wheat Germ Agglutinin Staining
The left ventricles of mice were collected and optimal cutting temperature compound embedded.They were then cut into 10 μm thick sections and stained separately with 5.0 μg/mL of WGA for 10 min at room temperature. Images were taken using an Olympus BX53 fluorescence microscope (Japan)equipped with a DP80 camera. Image-Pro Plus 6.0(Media Cybernetics, Bethesda, MD, USA) was used to calculate the cross-sectional areas. More than 60 cardiomyocytes in the examined sections were profiled in each group.
Transmission Electron Microscopy
Left ventricles (about 1 mm3) were collected from the same places for transmission electron microscopy (TEM) as described in previous studies.[19]The samples were fixed with a 2.5% glutaraldehyde solution and 1% osmic acid after being washed by phosphate-buffer saline (PBS). The samples were dehydrated, infiltrated, and finally embedded in epoxy resin. Slices were measured using an electron microscope (JEM-1200, JEOL Ltd., Tokyo, Japan). Digital images were obtained using support software packages. Morphological changes were described in diameter and roundness [(perimeter length)2/(4×π×area)] by using ImagePro-Plus 6.0 (Media Cybernetics, Bethesda, MD, USA).
Cell Culture and Treatment
H9c2 cells (ATCC, Manassas, VA, USA) were cultured in DMEM containing 10% fetal bovine serum,100 μg/mL streptomycin, and penicillin as described previously.[23]Trypsin digestion and cell subculture were performed after the cell fusion rate reached 80%. Cells were randomly divided into the control, ISO, 7,8-DHF-treated, and 7,8-DHF-only groups. In the ISO group, the cardiomyocyte hypertrophy model was induced by ISO at 10 μmol/L for 48 h as described in a previous study.[24]On the basis of our previous study,[19]7,8-DHF (100 μmol/L) was added 30 min before ISO treatment in the 7,8-DHF-treated group. 7,8-DHF (100 μmol/L, 48 h) was treated in the 7,8-DHF-only group. Treatment with Compound C (10 μmol/L) was performed for 12 h to validate the effect of AMPK. At the designated time, cells were collected for morphological or molecular biological experiments.
F-actin Staining
The Phalloidin-iFluor 488 staining kit was employed for staining actin filaments (F-actin) by using a previously reported method.[25]H9c2 cells cultured on the slide were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 30 min, and blocked with 2.5% bovine serum albumin for 30 min. The nucleus and actin filaments were visualized with DAPI (Beyotime, Haimen, China) and iFluor 488-conjugated phalloidin,respectively. Fluorescence images were acquired on a confocal laser scanning microscope (FV1000,Olympus, Japan). Image-Pro Plus 6.0 (Media Cybernetics, Bethesda, MD, USA) was used to measure the cell area.
ROS Detection
Cytoplasmic and mitochondrial ROS contents were examined using 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA, Beyotime, Haimen,China) or mito-SOX (Invitrogen, USA) in accordance with the protocols. Cells were treated as earlier described, trypsinized, and incubated with DCFH-DA (0.5 μmol/L) or mito-SOX (5 μmol/L) in PBS for 20 min at 37 °C in the dark. The images were acquired by laser scanning confocal microscopy (FV1000 or FV10i, Olympus, Japan).
Real-time PCR
Total RNA was extracted from H9c2 cells using TRIzol (Invitrogen). cDNA was synthesized from total RNA using a reverse transcription kit (Roche,Germany). SYBR Green I Master (Roche, Germany)was used for real-time polymerase chain reaction(PCR). The target genes were quantified using the LightCycle 480 Real-Time PCR system (Roche).Data were analyzed using theΔΔCt method. Primer sequences were as follows (5’-3’): ANP: F: GTGCGGTGTCCAACACAGAT; R: TCCAATCCTGTCAA TCCTACCC; brain natriuretic peptide (BNP): F:GAGGTCACTCCTATCCTCTGG; R: GCCATTTCCTCCGACTTTTCTC; β-myosin heavy chain (β-MHC): F: CCTGCGGAAGTCTGAGAAGG; R:CTCGGGACACGATCTTGGC, and GAPDH: F:CAAGGCTGAGAATGGGAAGC; R: GAAGACGCCAGTAGACTCCA.
Western Blot Analysis
Protein extraction and western blot detection of left ventricles and H9c2 cells were conducted as previously described.[20]The tissue/cell homogenate was centrifuged at 12,000 g for 30 min and the supernatants were collected for protein concentration detection using the BCA kit (Beyotime, Haimen,China). Protein samples were loaded and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to the nitrocellulose blotting membrane. Membranes were incubated with corresponding primary and secondary antibodies. The bands were visualized under the Odyssey Infrared Imaging System (LICOR Biosciences).Odyssey v2.1 was used for data analysis.
Statistical Analysis
All results were expressed as mean ± SE. The results were statistical analyzed using GraphPad Prism version 8.0 (GraphPad Software Inc., San Diego,CA, USA). The significance of the differences between the two groups was assessed by an unpaired Student'st-test. For multiple comparisons, one-way ANOVA followed by a Tukey’spost hoctest was used.The randomized block ANOVA (repeated measures ANOVA) was used for the data with a control value of 1 and no SE, as described previously.[26]P<0.05 was considered statistically significant.
RESULTS
Effects of 7,8-DHF on Decompensated Cardiac Hypertrophy
An 8-week TAC model simulating decompensated cardiac hypertrophy in mice was established to investigate the potential effects of 7,8-DHF on pressure overload-induced cardiac hypertrophy. The HW and TL were measured; HW was found to be significantly increased in the TAC mice, but decreased in the 7,8-DHF treatment mice (Figure 1A & 1B). By contrast, no statistical difference in TL was observed. HW/TL was significantly increased in the TAC mice but decreased in the 7,8-DHF-treated mice(Figure 1B). The cardiac function of TAC mice was also markedly impaired, as characterized by decreases in EF and FS, an increase in LVPW,d, and the tendency to increase in LVV,s and LVID,s. These alterations were reversed by 7,8-DHF. However, no significant difference was found in LVPW,s, LVV,d and LVID,d (Figure 1C & 1D). Therefore, 7,8-DHF inhibited decompensated cardiac hypertrophy.

Figure 1 7,8-DHF attenuated pressure overload-induced decompensate cardiac hypertrophy. (A): Representative pictures of murine hearts in the sham, TAC surgery for 8 weeks, and 7,8-DHF (5 mg/kg/d)-treated groups; (B): HW, TL, and HW/TL in the sham, TAC,and 7,8-DHF-treated mice; (C) representative images of echocardiography; (D): EF, FS, LVPW,s, LVPW,d, LVV,s, LVV,d, LVID,s, and LVID,d in sham, TAC, and 7,8-DHF-treated mice. Data are expressed as mean ± SE, n = 6 in each group. *P < 0.05, **P < 0.01 vs. sham, #P <0.05, ##P < 0.01 vs. TAC. EF: ejection fraction; HW: heart weight; FS: fractional shortening; LVID,s: end-systolic left ventricular internal diameter; LVID,d: end-diastolic left ventricular internal diameter; LVPW,s: left ventricular end-systolic posterior wall thickness;LVPW,d: left ventricular end-diastolic posterior wall thickness; LVV,d: end-diastolic left ventricular volume; LVV,s: end-systolic left ventricular volume; 7,8-DHF: 7,8-dihydroxyflavone; TAC: transverse aortic constriction; TL: tibia length.
Effects of 7,8-DHF on Compensated Cardiac Hypertrophy
In parallel, another TAC-induced cardiac hypertrophy mouse model was established for 4 weeks to simulate early-stage compensated hypertrophy. As shown in Figure 2A, murine hearts become enlarged after TAC operation, but are restored with 7,8-DHF. No significant difference in TL was found among these three groups. Meanwhile, HW and HW/TL were significantly increased in TAC mice but decreased in 7,8-DHF mice (Figure 2B). We further examined the cardiac function of mice by echocardiography (Figure 2C). Both EF and FS in TAC mice increased slightly relative to those in sham mice, which were reversed in 7,8-DHF-treated mice.Moreover, LVPW,s was increased, whereas LVV,s and LVID,s were decreased in TAC mice. These alterations were restored by 7,8-DHF. No significant difference was found in LVPW,d, LVV,d, and LVID,d(Figure 2D). These results indicate that TAC for 4 weeks elicited compensation of cardiac function,which was restored by 7,8-DHF. Collectively, these data suggest that 7,8-DHF impeded compensated and decompensated cardiac hypertrophy.
Effects of 7,8-DHF on Histology of Hypertrophic Myocardium
The histology and ultrastructure of murine hearts in each group were observed by HE staining, WGA staining, and TEM. As presented in Figure 3A & 3B,cardiac morphology disorders, together with histological injury, in TAC mice was found compared with that in sham mice, which was recovered in 7,8-DHFtreated mice. The protein level of the hypertrophic marker ANP was significantly increased in TAC but repressed by 7,8-DHF (Figure 3C). Moreover, 7,8-DHF significantly inhibited the cross-sectional area of TAC mice (Figure 3D). TEM examination suggested that the ultrastructure of mitochondria in TAC mouse ventricles was impaired, which was improved by 7,8-DHF. We further quantified the morphology of mitochondria in the ventricles. The diameter and roundness of mitochondria were decreased in TAC mice but restored by 7,8-DHF (Figure 3E).These findings indicate that ultrastructural abnormalities in mitochondria occurred earlier than functional deterioration. Moreover, 7,8-DHF attenuated mitochondrial disorders and the expression of hypertrophic markers, as well as preserved cardiac function in compensated cardiac hypertrophy.

Figure 2 7,8-DHF restored pressure overload-induced compensated cardiac hypertrophy. (A): Representative pictures of mouse hearts; (B): HW, TL, and HW/TL in sham, TAC surgery for 4 weeks and 7,8-DHF (5 mg/kg per day)-treated mice. N = 9 in each group.(C): Representative images of echocardiography. (D): EF, FS, LVPW,s, LVPW,d, LVV,s, LVV,d, LVID,s, and LVID,d in sham, TAC and 7,8-DHF-treated mice. N = 10 for the sham group, n = 13 for the TAC and +7,8-DHF groups. Data are expressed as mean ± SE. *P < 0.05 vs. sham, #P < 0.05, ##P < 0.01 vs. TAC. EF: ejection fraction; FS: fractional shortening; HW: heart weight; LVID,d: end-diastolic left ventricular internal diameter; LVID,s: end-systolic left ventricular internal diameter; LVPW,d: left ventricular end-diastolic posterior wall thickness; LVPW,s: left ventricular end-systolic posterior wall thickness; LVV,d: end-diastolic left ventricular volume; LVV,s: endsystolic left ventricular volume; 7,8-DHF: 7,8-dihydroxyflavone; TL: tibia length; TAC: transverse aortic constriction.
Effects of 7,8-DHF on the Expression of Mitochondria-related Factors in the Myocardium of Hypertrophic Mice
To further test which mitochondrial factor is acquired for the effects of 7,8-DHF, the expression levels of classical factors associated with mitochondrial function, including Mfn2, OPA1, Drp1, Fis-1,COXIV, and PGC-1α were determined. Among them,Mfn2 and OPA1 were identified as the key factors of mitochondrial fusion, whereas Drp1 and Fis-1 are the central factors of mitochondrial fission. COXIV expression significantly affected the functional state of the entire mitochondrial respiratory chain and cell energy production. PGC-1α is one of the critical regulators of mitochondrial biogenesis and energy metabolism.[27]The protein levels of Mfn2, OPA1, Drp1,Fis-1, and COXIV were not significantly affected among these three groups (Figure 4A-4E). However, PGC-1α was markedly decreased by TAC and restored by 7,8-DHF (Figure 4F).
Effects of 7,8-DHF on ISO-induced Cardiomyocyte Hypertrophy
To validate the antihypertrophic effects and potential mechanisms of 7,8-DHF, we established an ISO-induced cardiac hypertrophy model in H9c2 cells and further evaluated the effects of 7,8-DHF on the cell area and the expression of hypertrophic markers. Phalloidin staining of F-actin results showed that the area of ISO-treated cells was markedly larger than that of cells in the control group but reduced in 7,8-DHF-treated cells (Figure 5A). Meanwhile, transcriptional levels of classical hypertrophic marker genes,including ANP, BNP, and β-MHC, were detected to assess cardiac hypertrophy in H9c2 cells. As shown in Figure 5B, the mRNA levels of these three markers were significantly increased in ISO-treated cells but repressed by 7,8-DHF. Further, ISO-induced cytoplasmic ROS production in H9c2 cells was significantly reduced by 7,8-DHF (Figure 5C). Together,these findings suggest that 7,8-DHF inhibited cardiomyocyte hypertrophy and oxidative stress in ISOtreated H9c2 cells.

Figure 3 7,8-DHF relieved cardiac injury in cardiac hypertrophy mice. (A & B): Histological analyses of HE staining in the ventricles of mice in the sham, TAC surgery for 4 weeks, and 7,8-DHF (5 mg/kg per day)-treated groups; (C): representative western-blot bands and quantification of ANP, n = 6 in each group; (D): wheat germ agglutinin staining and quantification of average cross-sectional areas;18 pictures of 3 mice in each group; (E): transmission electron microscopy and quantification of the diameter and roundness of mitochondria in the ventricles of mice, 9 pictures of 3 mice in each group. Data are expressed as mean ± SE. **P < 0.01 vs. sham, ##P < 0.01 vs.TAC. 7,8-DHF: 7,8-dihydroxyflavone; TAC: transverse aortic constriction.
E ffects of 7,8-DHF on the Expression of Mitochondria-related Factors in ISO-induced H9c2 cells
Similarly, the protein expression of other mitochondrial dynamics-related molecules, including Mfn2,OPA1, and Drp1, were not significantly changed by 7,8-DHF (Figure 6A-6C). 7,8-DHF upregulated the protein expression of PGC-1α in ISO-treated H9c2 cardiomyocytes (Figure 6D).
Essential Role of AMPK in the Anti-hypertrophic Effects of 7,8-DHF
AMPK is a central regulator of mitochondrial function and energy metabolism; therefore, we detected the expression and activity of AMPK and downstream STAT3 signal, which are closely involved in the development of cardiac hypertrophy. We found that 7,8-DHF markedly elevated the protein level of p-AMPK but decreased p-STAT3 in ISO-treated H9c2 cells (Figure 7A & 7B). A specific AMPK inhibitor was employed to evaluate whether AMPK is necessary for the effects of 7,8-DHF. AMPK inhibition recovered the cell area of cardiomyocytes (Figure 7C).We also found that AMPK inhibition markedly eliminated the antioxidative role of 7,8-DHF by increasing the production of mitochondrial ROS (Figure 7D).Overall, these findings suggest that 7,8-DHF inhibited cardiac hypertrophy and related mitochondrial oxidative stress by activating AMPK signaling.
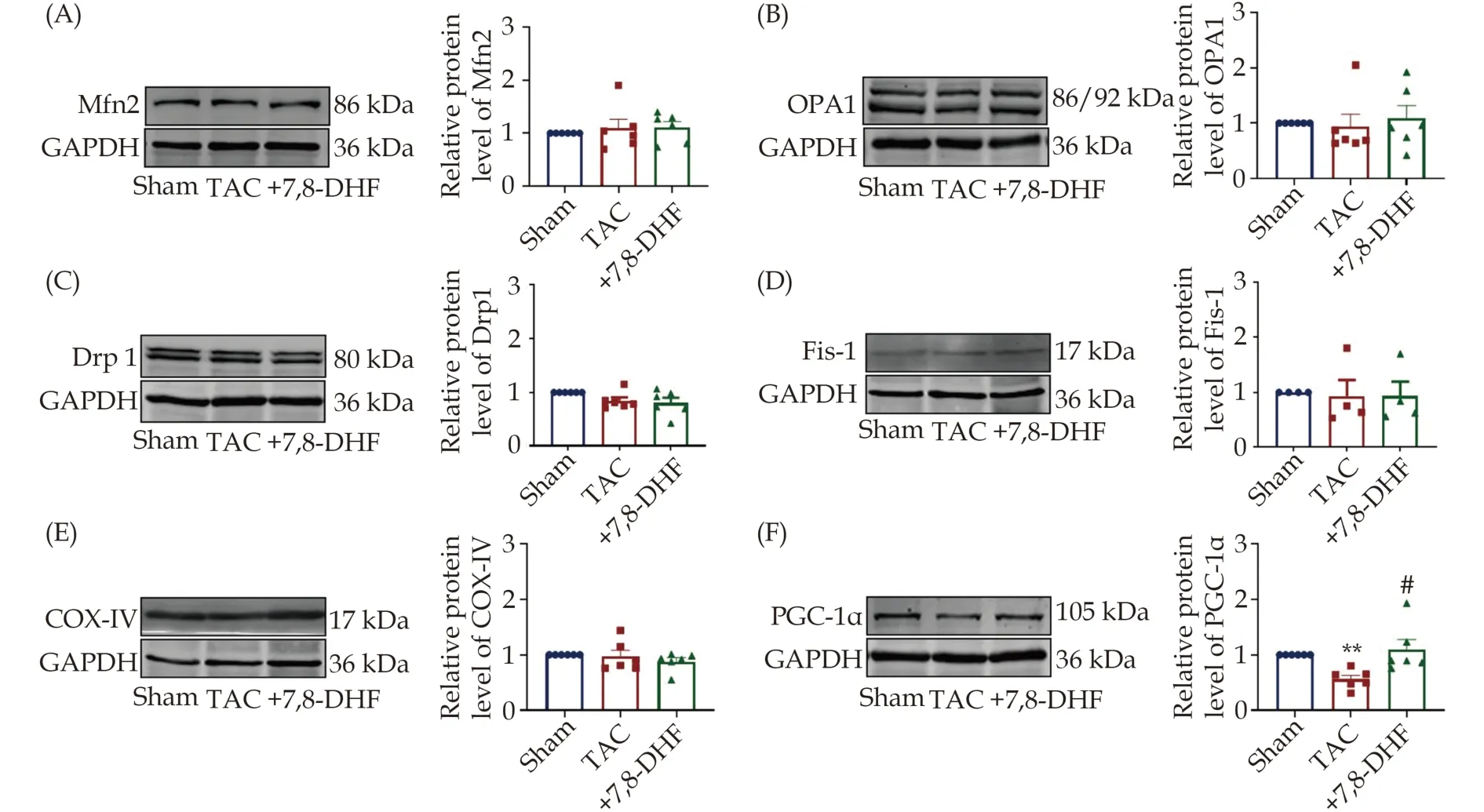
Figure 4 Protein expression of mitochondria-related factors in the ventricles of 7,8-DHF-treated hypertrophic mice. (A-F): Representative Western blots (left) and quantification (right) of mitochondria-related factors, including Mfn2, OPA1, Drp1, Fis-1, COX-IV, and PGC-1α in left ventricles from the sham, TAC for 4 weeks, and 7,8-DHF-treated groups. Data are expressed as mean ± SE. N = 4 the in sham group; n = 6 in the TAC, and +7,8-DHF groups for Mfn2, OPA1, and Drp1; and n = 4 in each group for Fis-1. Data are expressed as mean ± SE. **P < 0.01 vs. sham, #P < 0.05 vs. TAC. 7,8-DHF: 7,8-dihydroxyflavone; TAC: transverse aortic constriction.
DISCUSSION
The transition from cardiac hypertrophy to heart failure is a dynamic process comprising the compensated stage at the early phase of pressure overload and the advanced decompensated stage after long-term stimuli.[28]Accumulating evidence supported the view that mitochondrial dysfunction is significantly involved in decompensated cardiac hypertrophy.[29]In the current study, we evaluated the effects of the small-molecule BDNF mimetic 7,8-DHF on both compensated and decompensated cardiac hypertrophy. This study mainly found that 7,8-DHF inhibited the progression of mitochondrial dysfunction by activating the AMPK/PGC-1α signaling pathway, which provided a novel pharmacological effect and the action mechanism of 7,8-DHF in cardiac hypertrophy (Figure 8).
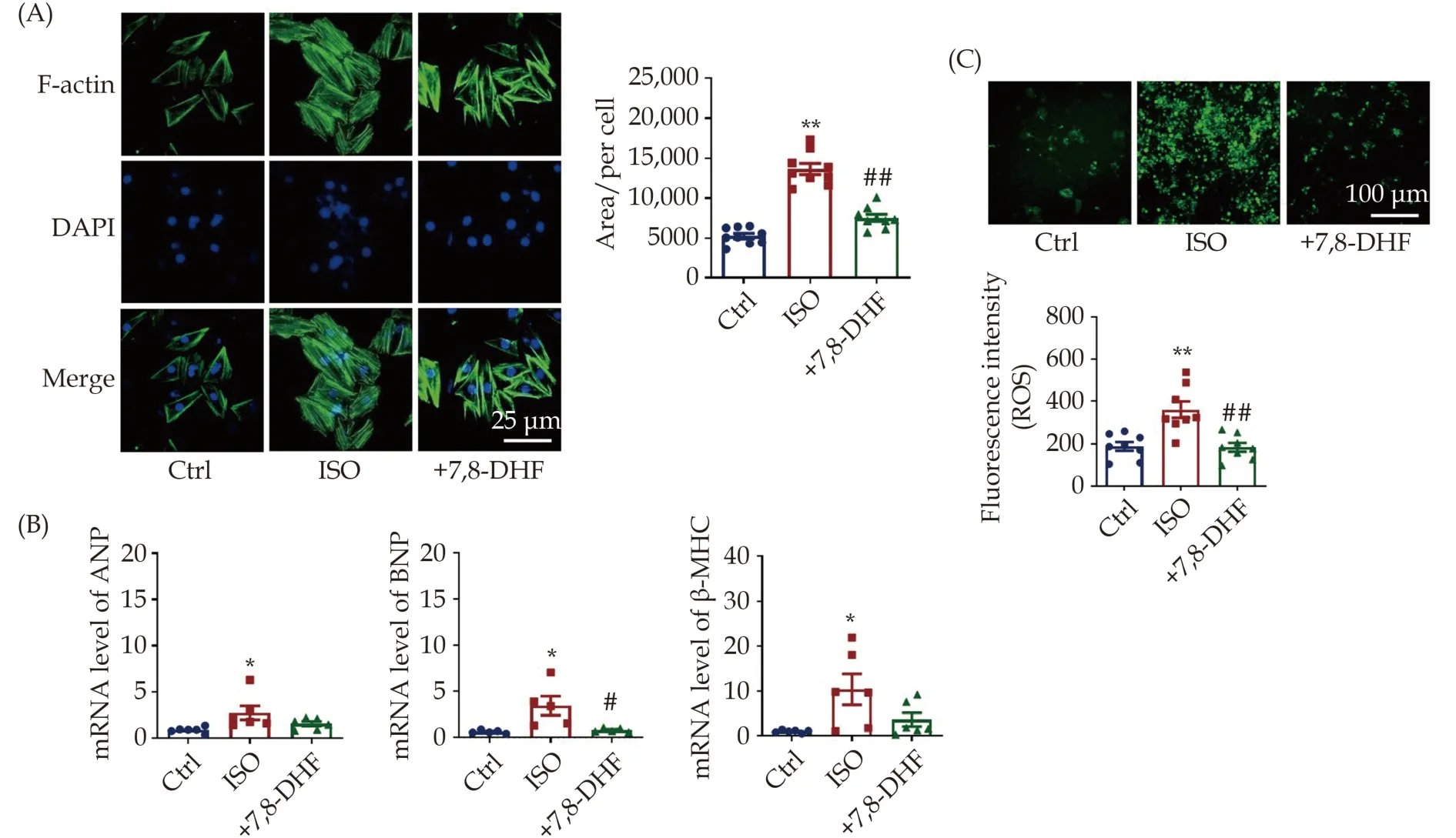
Figure 5 7,8-DHF inhibited isoproterenol-induced cardiomyocyte hypertrophy in H9c2 cells. (A): The cardiomyocyte hypertrophy model was induced by ISO (10 μmol/L) for 48 h in H9c2 cells. 7,8-DHF (100 μmol/L) was pretreated 30 min before ISO administration.The cell skeleton and nucleus were stained using the Phalloidin-iFluor 488 reagent or DAPI. Green fluorescence indicates actin filaments, whereas blue fluorescence indicates the nuclei. N = 9 in each group; scale bar: 50 μm. (B): The transcript levels of three hypertrophic marker genes including ANP, BNP, and β-MHC. N = 6 in each group for ANP and β-MHC, n = 5 in each group for BNP. (C):Cytoplasmic reactive oxygen species were stained with a DCFH-DA probe. Scale bar: 100 μm, n = 8 in each group. Data are expressed as mean ± SE. *P < 0.05, **P < 0.01 vs. control, #P < 0.05, ##P < 0.01 vs. ISO. ISO: isoproterenol; 7,8-DHF: 7,8-dihydroxyflavone.

Figure 6 Protein expression of mitochondria-related factors in 7,8-DHF-treated hypertrophic H9c2 cells. (A-D): Representative Western blots and quantification of mitochondria-related key factors, including Mfn2, OPA1, Drp1, and PGC-1α in H9c2 cells. Data are expressed as mean ± SE, n = 6 in each group. **P < 0.01 vs. control, #P < 0.05 vs. ISO. ISO: isoproterenol; 7,8-DHF: 7,8-dihydroxyflavone.

Figure 7 Adenosine monophosphate-activated protein kinase plays an important role in the antihypertrophic effect of 7,8-DHF.(A): Representative Western blots analysis and the quantification of AMPK and its phosphorylation in ISO-induced H9c2 cells. N = 6 in each group. (B): Representative Western blot bands and quantification of p-STAT3/STAT3. N = 6 in each group. (C): Effect of compound C (10 μmol/L, 12 h) on the antihypertrophic role of 7,8-DHF in ISO-induced H9c2 cells. N = 5 in each group. (D): Effect of compound C on the antioxidative role of 7,8-DHF in ISO-induced H9c2 cells. Red fluorescence indicates mitochondrial ROS, whereas blue fluorescence indicates nuclei. Scale bar: 10 μm, n = 6 in each group. Data are expressed as mean ± SE. *P < 0.05, **P < 0.01 vs. control, #P< 0.05, ##P < 0.01 vs. ISO, &P < 0.05, &&P < 0.01 vs. +7,8-DHF. ISO: isoproterenol; 7,8-DHF: 7,8-dihydroxyflavone.
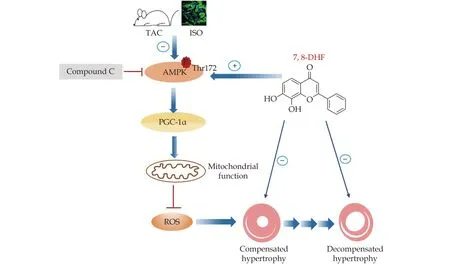
Figure 8 Schematic of the protective effects of 7,8-DHF against cardiac hypertrophy via the activation of AMPK/PGC-1α signaling.The in vivo and in vitro experiments show that the small-molecule BDNF mimetic 7,8-DHF inhibits compensated and decompensated cardiac hypertrophy. Treatment with 7,8-DHF promotes AMPK phosphorylation and the upregulation of PGC-1α expression, thus contributing to the preservation of cardiac function and mitochondrial homeostasis, as well as the alleviation of oxidative stress in TAC/ISO-induced cardiomyocytes. However, the administration of the AMPK inhibitor Compound C blocks the effects of 7,8-DHF.ISO: isoproterenol; 7,8-DHF: 7,8-dihydroxyflavone.
Many previous studies employed the TAC model for 4 or 8 weeks to establish pathological cardiac hypertrophy.[30,31]Among them, TAC for 4 weeks usually reflects the early phase of cardiac hypertrophy(cardiac function increases or slightly decreases), whereas TAC for 8 weeks or longer indicates the late phase (cardiac function markedly decreases). Similarly,previous studies have suggested that cardiac function is preserved in mice after TAC operation for 3 or 5 weeks.[32,33]In our study, we established 4-week TAC-induced compensated hypertrophy and 8-week TAC-induced decompensated hypertrophy. Oxidative stress is known to be closely associated with ISOinduced cardiomyocyte hypertrophy.[34,35]In the present study, treatment with 7,8-DHF significantly inhibited both cytoplasmic and mitochondrial ROS,suggesting that the elimination of oxidative stress was required. Previous studies, including the research conducted by our group, have associated 7,8-DHF with mitochondrial function and energy homeostasis. For example, 7,8-DHF increases mitochondrial respiratory capacity by activating the biogenesis activator PGC-1α and CREB phosphorylation in neuroblastoma cells.[36]Similarly, 7,8-DHF attenuates traumatic brain injury and promotes early brain tra
uma recovery by restoring PGC-1α and AMPK-mediated energy metabolism.[37,38]The critical role of 7,8-DHF in the energy metabolism of skeletal muscle has also been reported. The anti-obesity effect of 7,8-DHF on female mice on a high-fat diet is found by increasing systemic energy expenditure.[39]Further research by this group has confirmed that 7,8-DHF promotes mitochondrial biogenesis by activating the AMPK/CREB/PGC-1α pathway in cultured skeletal muscle cells.[40]Specifically, two other studies suggested that BDNF protected against skeletal atrophy and improved the exercise capacity of mice with heart failure by activating AMPK/PGC-1α signaling.[16,41]Consistently, the present study indicated that 7,8-DHF restrained cardiac hypertrophy and heart failure by activating AMPK/PGC-1α in cardiomyocytes. Our previous report documented that 7,8-DHF increased mitochondrial oxidative phosphorylation and inhibited excessive mitochondrial fission in doxorubicin-induced cardiomyopathy and myocardial ischemic mice, respectively.[19,20]By contrast, 7,8-DHF exerted no significant effect on mitochondrial dynamics-related key molecules in this study. These differences may be associated with the pathogenesis of different experimental models.In addition, the response of AMPK, the sensor in energy metabolism, is more sensitive than other factors.[42]Notably, 7,8-DHF inhibited AMPK activity in doxorubicin or ischemia-induced cardiac injury in our previous studies,[19,20]but activated AMPK in cardiac hypertrophy. This discrepancy may be caused by the two sides of AMPK and its dominant pathway. Thus, 7,8-DHF not only improves mitochondrial dysfunction by activating AMPK/PGC-1α but also potentially inhibits the imbalance of mitochondrial dynamics and cardiotoxicity by downregulating the overactivation of AMPK signals.[19]Collectively, a dynamic regulation exists between the 7,8-DHF and AMPK-mediated pathways.
In the current study, treatment with 7,8-DHF only highly activated AMPK. Previous studies have verified that 7,8-DHF alleviates hyperlipidemia, hyperglycemia, and lipid accumulation in the skeletal muscle and liver of obese animals.[40]Accordingly, whether 7,8-DHF exhibits similar pharmacological properties to those of metformin is interesting to explore. Whether 7,8-DHF has potential adverse effects should also be determined. The cytotoxicity of chronic treatment with 7,8-DHF has been evaluated in previous studies; no adverse pathological change has been detected in drug-treated viscera organs and complete blood count.[43]Moreover, in our study, treatment with 7,8-DHF for 8 weeks has no effect on heart function under physiological conditions (data not shown).AMPK activation requires phosphorylation by upstream kinases such as liver kinase B1 (LKB1) and calcium/calmodulin-dependent protein kinase kinase-β of a threonine residue (Thr172). In addition,numerous signaling pathways have been revealed to be linked to AMPK as downstream targets, including both metabolic and non-metabolic signaling. The signaling pathway mainly controlled by AMPK remains unclear, although PGC-1α expression is found increased, accompanied by activated AMPK. Thus,further research will be conducted to fully clarify the detailed mechanisms of 7,8-DHF in activating AMPK signals.
In the current study, we found that decompensation induced by treatment with TAC for 8 weeks was restored by 7,8-DHF. Notably, compensation induced by treatment with TAC for 4 weeks was reversed in 7,8-DHF-treated murine hearts, suggesting that 7,8-DHF may also inhibit physiological compensation. A similar phenomenon has been previously reported.[44]Actually, mitochondrial injury has been observed in cardiomyocytes, although the cardiac function is compensated. This finding was supported by the activation of STAT3, a pro-hypertrophic factor. However, 7,8-DHF recovered mitochondrial dysfunction and the activity of STAT3, accompanied by the restoration of cardiac function. Thus, cardiac function was speculated to provide protection owing to the inhibition of oxidative stress and mitochondrial disorders in 7,8-DHF-treated mice. In this case, the cardiomyocytes do not need to grow to overcome pathological insults such as pressure overload.The concrete causal link among them requires further investigation in the subsequent work.
The present study has limitations. First, the pressure-overload mouse model was used in this study,and whether 7,8-DHF attenuated the development of cardiac hypertrophy induced by other approaches is unknown. However, the cellular study suggested that 7,8-DHF could also be repressed cardiomyocyte hypertrophy in ISO-induced H9c2 cells. Meanwhile,the left and right ventricles may vary in performance,particularly in long-time cardiac hypertrophy/heart failure, which should also be investigated in the subsequent study. Second, the H9c2 cardiomyoblast cell line instead of primary neonatal cardiomyocytes were cultured and stimulated in thein vitrosystem.An early study suggested that H9c2 cells displayed similar hypertrophic responses to neonatal primary cardiomyocytes.[45]The regulatory role of 7,8-DHF should also be validated in primary neonatal cardiomyocytes and other cardiac cell lines. Third, the concrete mechanisms of 7,8-DHF on energy metabolism in pathological cardiac hypertrophy by regulating AMPK and mitochondrial function need to be further explored.
CONCLUSIONS
The present study is the first to demonstrate that 7,8-DHF is an important candidate to counteract the compensation and decompensation of cardiac hypertrophy by targeting AMPK signaling. These findings provide a novel insight into the pharmacological effect of 7,8-DHF in heart diseases and the treatment of diseases induced by mitochondrial dysfunction.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (No. 82070309, 8187 0191, and 82073838), “333 Project” of Jiangsu Province and the Lvyangjinfeng Talent Program of Yangzhou.
AUTHOR CONTRIBUTIONS
Peng-Zhou HANG and Jing ZHAO: conception and design, analysis and interpretation of data; Pei-Feng LI, Jie LIU, Feng-Feng LI, Ting-Ting CHEN and Yang PAN: performance of experiments; Peng-Zhou HANG,Jing ZHAO, Zhi-Min DU, Hong-Yu JI, Man-Ru ZHANG,and Hua-Qing YU: drafting of the manuscript or revising it critically for important intellectual content;Zhi-Min DU and Jing ZHAO final approval of the manuscript submitted.
CONFLICTS OF INTEREST
None.
杂志排行

Journal of Geriatric Cardiology的其它文章
- Review on the management of cardiovascular risk factors in the elderly
- Observation of performance measures of STEMI in elderly after implementation of an updated protocol: results from a single center without coronary interventions
- Migration of intra-aortic balloon pump causing obstruction of the superior mesenteric artery
- Device-based neuromodulation for cardiovascular diseases and patient’ s age
- Methodology in coronary artery bypass surgery quality assessment
- Validating the accuracy of a multifunctional smartwatch sphygmomanometer to monitor blood pressure