紫甘薯水溶性多糖的提取工艺优化及结构研究
2022-12-20李晨京冯怡华王春玲
李晨京,冯怡华,王春玲
(天津科技大学 食品科学与工程学院,天津 300457)
紫甘薯,属于旋花科甘薯属,是一种双子叶农业作物[1]。它在世界的三大温度带(热带、副热带和温带)地区种植广泛,因其产量高和对不同环境、温度和土壤条件的适应能力强,紫甘薯被认为是甘薯市场上最有优势的经济作物之一[2]。我国于20世纪90年代引进紫甘薯,并在河北、河南、山东、江苏等地种植[3]。紫甘薯营养丰富,与普通甘薯相比,其含有更多的氨基酸、淀粉、可溶性糖、花青素等[4]。近几年,紫甘薯以其特有的营养价值受到越来越多人的喜爱,因此其种植面积也呈现出逐年增加的趋势,市场开发潜力很大。
植物多糖是一种具有生物活性的大分子,由相同或不同数量的单糖组成,研究发现多糖具有抗炎、预防癌症、抗氧化、降低血糖、保护肝脏[5-8]等多种生物活性,是保健类食品开发的重要原料之一。目前能够参与体内生理功能的天然多糖已被鉴定出300多种[9],且大部分植物多糖都比较安全,不会产生明显的副作用[10]。多糖提取方法有很多,酸法和碱法提取多糖时,由于提取液的酸碱度比较高,容易引起糖苷键断裂,破坏多糖结构[11-12]。超声波辅助提取和微波辅助提取虽然可以比较彻底地破坏细胞壁,提高多糖的提取率,但也会对多糖的结构产生破坏作用[13]。而热水提取是在一定的温度和时间内提取多糖[14],它因具有成本低、操作简单、试剂无污染、提高多糖的溶解度等优点而备受青睐。近年来对于紫甘薯,国内外的学者的研究主要集中于对其花青素[15]、黄酮类化合物[16]、酚酸衍生物[17]等具有活性成分的物质分离,但是对紫甘薯多糖的提取、工艺的优化以及结构鉴定的研究鲜见。
因此,本试验以紫甘薯为原料,采用热水提取法提取多糖,通过单因素和响应面试验优化提取工艺,并对紫甘薯多糖进行分离纯化和结构分析鉴定,为后续的开发利用提供基础。
1 材料与方法
1.1 材料与试剂
紫甘薯(紫罗兰品种):市售。
石油醚、无水乙醇、正丁醇、浓硫酸、三氟乙酸、甲醇(均为分析纯):天津市化学试剂一厂;三氯甲烷(分析纯):上海蓝季科技公司;重蒸酚、标准葡聚糖(分析纯)、Sepharose 4B、α-淀粉酶(≥40 000 U/g,食品级)、糖化酶(≥100 000 U/g,食品级)、蛋白酶(≥100 000 U/g,食品级)、鼠李糖、阿拉伯糖、半乳糖、葡萄糖、木糖、甘露糖、葡萄糖醛酸、半乳糖醛酸(均为分析纯):北京Sorlabio生物公司。
1.2 仪器与设备
多功能粉碎机(1500C):东莞市华泰电器有限公司;Freezone冻干机(6 plus):美国 Labconco公司;精密天平(Q224-1CN):美国Sartorius公司;磁力搅拌器(Tex-Ⅱ)、高性能台式离心机(X1R)、酶标仪(MULTISKAN GO)、高效阴离子色谱仪(ICS-5000+):美国Thermo公司;电热恒温水浴锅(BCI-HW):上海医疗器械五厂;旋转蒸发器(3000A)、自动部分收集器(BSZ-100):上海亚荣生化仪器厂;蠕动泵(100M):保定创锐泵业有限公司;高效液相色谱仪(LC20A):日本岛津公司;紫外分光光度计(U-T6):上海屹谱仪器制造有限公司;傅里叶变换红外光谱检测仪(IS50):美国布鲁克仪器公司。
1.3 试验方法
1.3.1 紫甘薯预处理
紫甘薯洗净、去皮,切成5 cm×0.5 cm×0.5 cm的矩形长条,-80℃冻干机中冷冻干燥24 h至无水分,粉碎过筛后装袋储存在干燥器中。
1.3.2 单因素试验
1.3.2.1 提取温度对紫甘薯多糖提取率的影响
每份称取10 g紫甘薯粉末,共计5份。在设定提取时间 2.5 h、液料比为 20∶1(mL/g)的条件下,于 50、60、70、80、90℃的温度下提取多糖,离心收集上清液并浓缩。水浴锅60℃加热浓缩液,加入适量糖化酶水解30 min后温度调至95℃,加入适量淀粉酶水解15 min,冷却至室温(26℃)。结束后,缓慢加入浓缩液体积4倍的无水乙醇,4℃冷藏12 h,离心后除去上清,加入适量蒸馏水复溶。采用蛋白酶-Sevag联合法除蛋白,首先水浴锅70℃加热后,加入适量蛋白酶水解30 min后冷却;其次加入1/4多糖溶液体积的Sevag试剂,摇床均匀振荡15 min离心除去白色沉淀物,重复此操作6次。AB-8大孔吸附树脂脱除色素,作用时间为12 h,结束后用蒸馏水冲洗3次树脂以充分获得全部多糖。除去色素的多糖溶液浓缩至1/10体积,冻干后得到紫甘薯多糖。测定糖含量,得出提取率。
1.3.2.2 提取时间对紫甘薯多糖提取率的影响
每份称量10 g紫甘薯粉末,共计5份。设定提取温度80℃、液料比为20∶1(mL/g)恒定不变,提取时间分别取 1.0、1.5、2.0、2.5、3.0 h。其余操作步骤同 1.3.2.1。
1.3.2.3 液料比对紫甘薯多糖提取率的影响
每份称量10 g紫甘薯粉末,共计5份。设定提取温度80℃、提取时间为2.5 h恒定不变,液料比分别取5∶1、10∶1、15∶1、20∶1、25∶1(mL/g)。其余操作步骤同1.3.2.1。
1.3.3 紫甘薯多糖含量的测定
采用苯酚-硫酸法[18]测定紫甘薯中的多糖含量。
1.3.3.1 标准曲线的绘制
精确称取干燥无水的葡萄糖10.0 mg,使用100 mL容量瓶定容,即得到0.1 mg/mL的标准葡萄糖溶液。分别吸取 0、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9 mL 于各试管中,蒸馏水补足至终体积为1.0 mL。向各管加入5.0 mL浓硫酸与1.0 mL 6%苯酚,均匀振荡后加热煮沸15 min,冷却后于490 nm测定吸光值。
1.3.3.2 紫甘薯多糖含量的测定
称取一定质量的多糖,定容至250 mL,依照标椎曲线的方法加入试剂进行试验,并以此计算出紫甘薯多糖的提取率。紫甘薯多糖提取率的计算如下。

式中:R为紫甘薯多糖的提取率,%;C为紫甘薯多糖浓度,μg/mL;V为样液体积,mL;D为稀释倍数;W为所称样品的质量,g。
1.3.4 响应面试验
响应面试验因素与水平见表1。
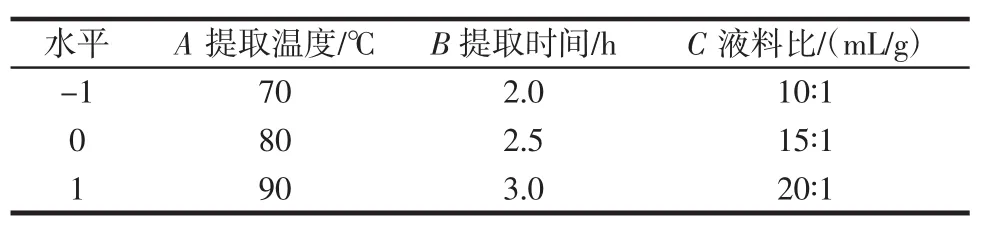
表1 响应面试验因素与水平Table 1 Response surface experiment factors and levels
1.3.5 紫甘薯多糖的分离纯化
Sepharose 4B的特点是分子量分离范围较大,根据它的特点选用其作为层析的填料进行紫甘薯多糖的分离和纯化,方法参考文献[19]并进行适当修改。
将紫甘薯粗多糖用超纯水配制成20mg/mL的多糖溶液,过0.22μm水系滤膜后在Sepharose4B(id1.6cm×60 cm)凝胶层析柱中上样2.0 mL,以超纯水洗脱,流速设置为0.5 mL/min,每管的收集时间设定为4 min,用苯酚-硫酸法检测各管吸光值,将收集管数作为横坐标,各管的吸光度值作为纵坐标,绘制紫甘薯多糖的洗脱曲线。根据曲线将洗脱峰对应管数分别合并收集,低温真空冷冻干燥,得纯化后多糖组分。
1.3.6 多糖的分子量测定
采用高效液相色谱法进行紫甘薯多糖的纯度鉴定。精确称量标准葡聚糖各5.0 mg,分子量分别为4×104、1×105、2×105、5×105、1×106、2×106。1 mL 超纯水溶解后,过0.22 μm水系膜除杂,进样针吸取20 μL进样。根据不同分子量葡聚糖的出峰时间制作标准曲线,得到回归方程。
纯多糖按照上述方法进行溶解,过膜后上样,采用高效液相色谱法进行检测,根据保留时间按照回归方程计算分子量。
1.3.7 紫甘薯多糖的纯度鉴定
紫外分光光度计在200 nm~400 nm处扫描浓度为1.0 mg/mL的纯多糖溶液。基于260 nm和280 nm处曲线是否光滑来判定多糖中是否存在核酸和蛋白质[20]。
1.3.8 红外光谱分析
首先称量100 mg干燥的KBr,研磨成粉末后模具加压1 min制成近乎透明的薄片,红外光谱仪在4 000 cm-1~400 cm-1内进行扫描,测定空白背景。1 mg干燥至恒重的纯多糖与100 mg KBr混合后迅速研磨至粉状,制片后采用相同方法扫描并分析多糖的特征吸收峰。
1.3.9 单糖组成分析
1.3.9.1 单糖标准品的配制
称量适量的8种单糖标准品(鼠李糖、阿拉伯糖、半乳糖、葡萄糖、木糖、甘露糖、半乳糖醛酸、葡萄糖醛酸)于试管中,超纯水溶解配制成1 mg/mL的混合单糖标准液,4℃冰箱储存备用。
1.3.9.2 样品处理
试管中加入5 mg的纯多糖,N2环境下加入5 mL的三氟乙酸(2 mol/L),确保N2充满整个试管后迅速封管。120℃的油浴锅中降解反应3 h,取出冷却至室温。50℃蒸干样品后加入1 mL甲醇继续蒸干,操作重复5次。加入5 mL超纯水振荡溶解样品,用0.22 μm水系滤膜、小柱过滤后,吸取1 mL准备上样。
1.3.9.3 流动相的配制
200 mmol/L NaOH溶液:4.2 mL NaOH溶液定容至400 mL,摇匀,通N2保护备用。
1mol/L醋酸钠溶液:称取8.2g醋酸钠固体,用50mL超纯水溶解,减压抽滤后定容至100 mL,摇匀后通N2保护备用。
800 mL超纯水,通N2保护备用。
1.3.9.4 检测条件
Thermo Dionex ICS2500色谱系统,色谱柱为Carbo Pac PA10(150 mm×3 mm),柱温设为 30℃,进样量1 mL,检测时间为40 min。
根据8种单糖的色谱图出峰时间与纯糖进行分析比较,对纯多糖的单糖组成进行定性,结合峰面积、相对分子质量等其他数据计算出纯糖中各单糖的摩尔比。
1.4 数据分析与处理
利用Design-Expert.V8.0和Origin 8.6等软件对数据进行分析及制图。
2 结果与分析
2.1 紫甘薯多糖提取的单因素试验
2.1.1 提取温度对紫甘薯多糖提取率的影响
提取温度对紫甘薯多糖提取率的影响结果如图1所示。
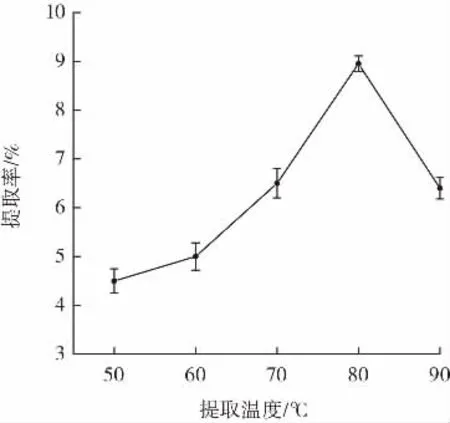
图1 不同提取温度对多糖提取率的影响Fig.1 Influence of different extraction temperature on extraction yield of polysaccharides
由图1可知,紫甘薯多糖提取的温度较低时,随着提取温度的升高,多糖提取率随之增加,说明了温度升高可促进细胞内多糖分子的运动,以促进多糖溶出[21],当温度升高到80℃时,多糖提取率最大。但当温度升高到90℃时,糖苷键可能会断裂,发生多糖降解。通过分析以上结果,选取提取温度80℃作为紫甘薯多糖的最佳提取温度。
2.1.2 提取时间对紫甘薯多糖提取率的影响
提取时间对紫甘薯多糖提取率的影响结果如图2所示。
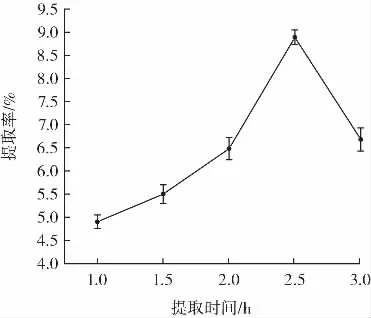
图2 不同提取时间对多糖提取率的影响Fig.2 Influence of different extraction time on extraction yield of polysaccharides
由图2可知,当提取时间设定在1.0 h~2.5 h内时,随着时间的逐渐延长,紫甘薯多糖的提取率也逐渐增加,并在2.5 h时达到最大,说明了提取时间适当的延长也可提高多糖的提取效率[22]。但过长的时间,多糖的溶解逐渐达到平衡后,可能会引起多糖结构的改变从而降低提取率。因此选定2.5 h为紫甘薯多糖的最优提取时间。
2.1.3 液料比对紫甘薯多糖提取率的影响
液料比对多糖提取率的影响结果如图3所示。
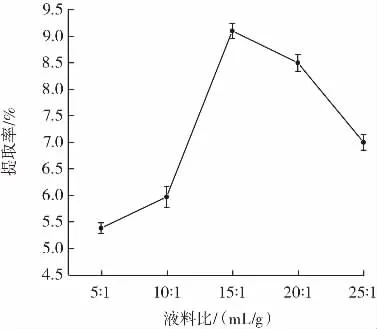
图3 不同液料比对多糖提取率的影响Fig.3 Influence of different liquid-solid ratio on extraction yield of polysaccharides
由图 3 可知,在液料比为 15∶1(mL/g)时,多糖得率最大。当紫甘薯细胞内多糖浸出率达到最大值之前,多糖的提取率随着溶剂用量的增大而变大,但当多糖在溶液中几乎完全浸出后,过多的溶剂使后续步骤更加繁琐,引起大量多糖被损耗,从而提取效率下降[23]。因此选取15∶1(mL/g)为紫甘薯多糖提取的最佳液料比。
2.2 紫甘薯多糖提取的响应面试验
2.2.1 响应面试验设计
通过对单因素的试验结果进行分析后,确定了提取温度:70、80、90 ℃,提取时间:2.0、2.5、3.0 h,提取液料比 10∶1、15∶1、20∶1(mL/g)作为下一步试验因子。响应值为多糖的提取率,响应面试验使用Design-Expert 8.0.6软件进行设计,试验方案共17组,试验后通过计算得出多糖提取率,结果见表2。
2.2.2 拟合模型与显著性检验
应用Design-Expert 8.0.6软件对表2中的数据进行了建模和分析,得到了提取率(R)的提取温度(A)、提取时间(B)、液料比(C)的多元回归方程:R/%=9.00+0.23A+0.33B+0.34C-0.65AB-0.041AC-0.23BC-1.58A2-1.16B2-1.60C2。
响应面试验数据的分析结果见表3。
由表3可知,P<0.000 1,R2=0.995 6,说明此模型显著性很高;且失拟项P值0.728 3>0.05,说明明显的失拟因素不存在,因此该回归方程可信。F检验可评估判断自变量对因变量产生的影响[24],可知3个因素影响提取率的程度为C(液料比)>B(提取时间)>A(提取温度)。
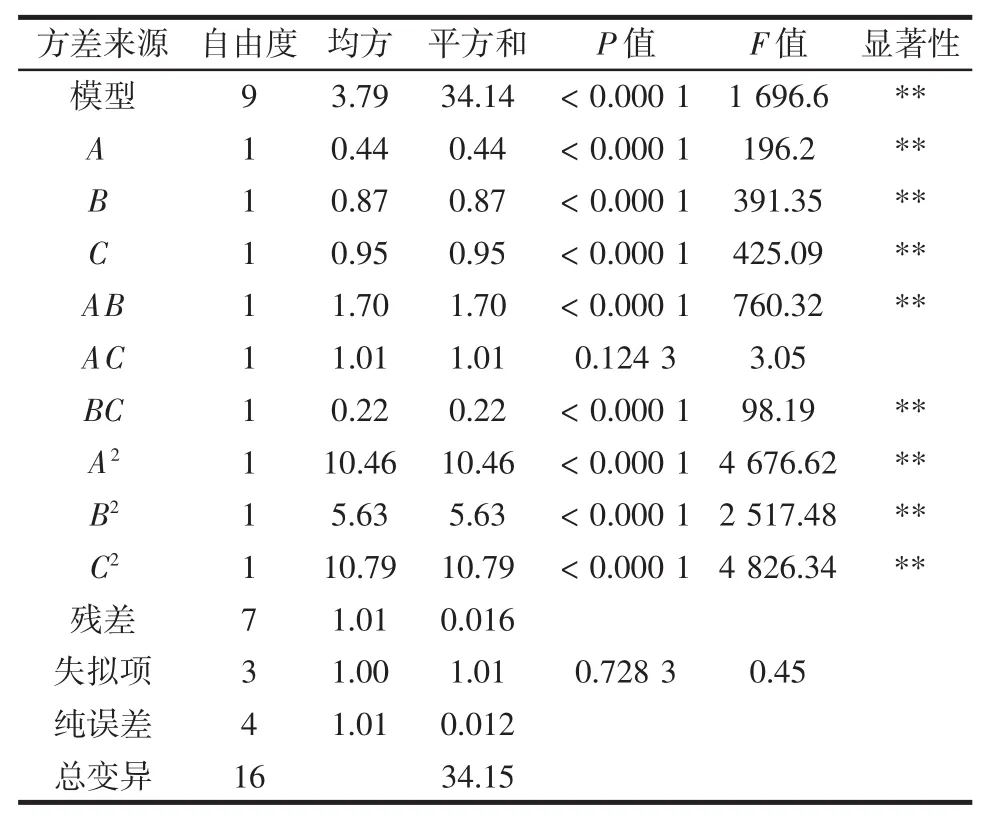
表3 Box-Behnken数据分析结果Table 3 Results of Box-Behnken data analysis
2.2.3 响应面试验结果分析
3个因素的两两交互作用对提取率的影响见图4~图6。等高线若为圆形则表明两因素间的相互作用对提取率的影响很小,若是椭圆形则说明影响很大[25]。
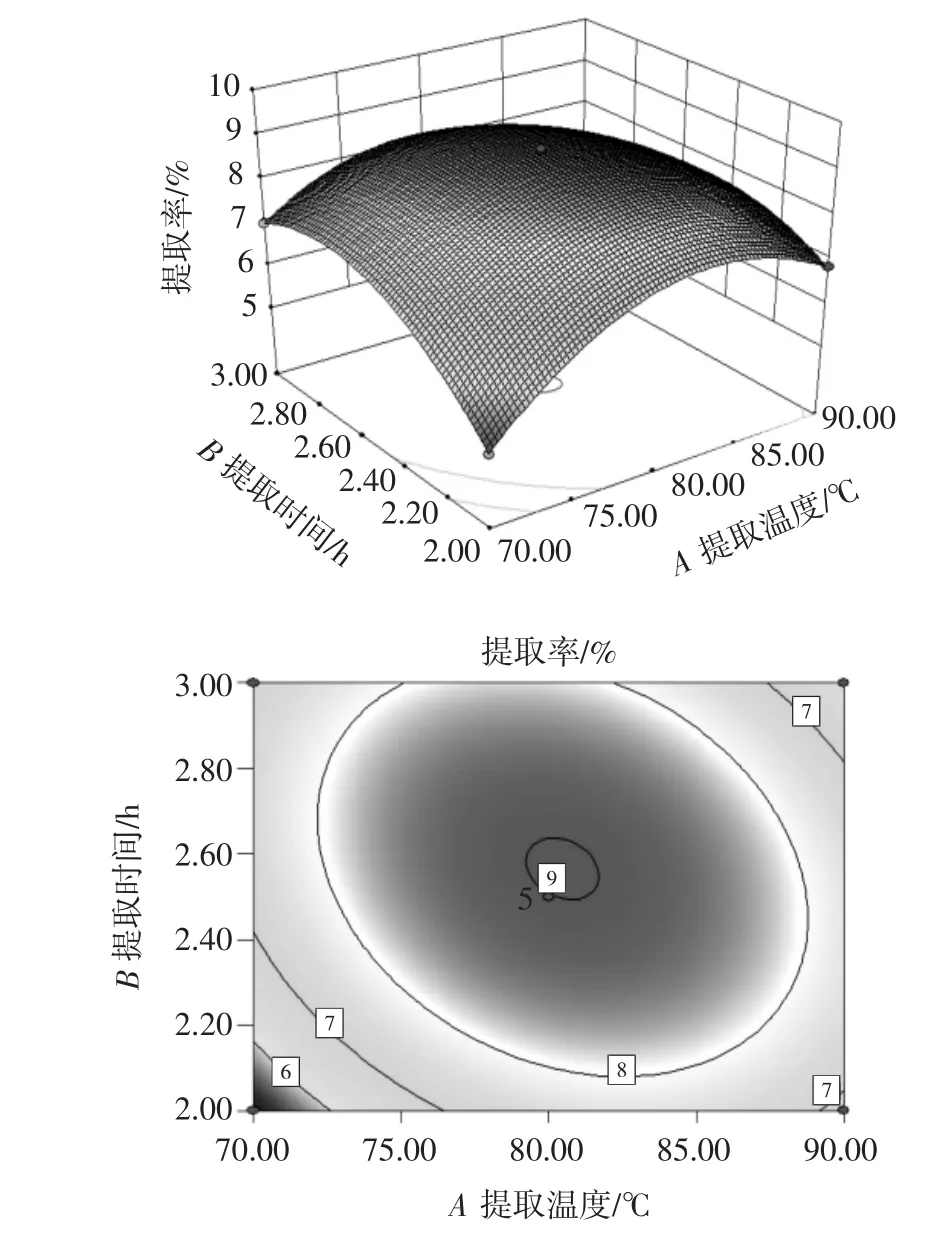
图4 提取温度和提取时间两因素对提取率的交互作用Fig.4 Interaction of extraction temperature and extraction time on extraction yield

图5 提取温度和液料比两因素对提取率的交互作用Fig.5 Interaction of extraction temperature and liquid-solid ratio on extraction yield
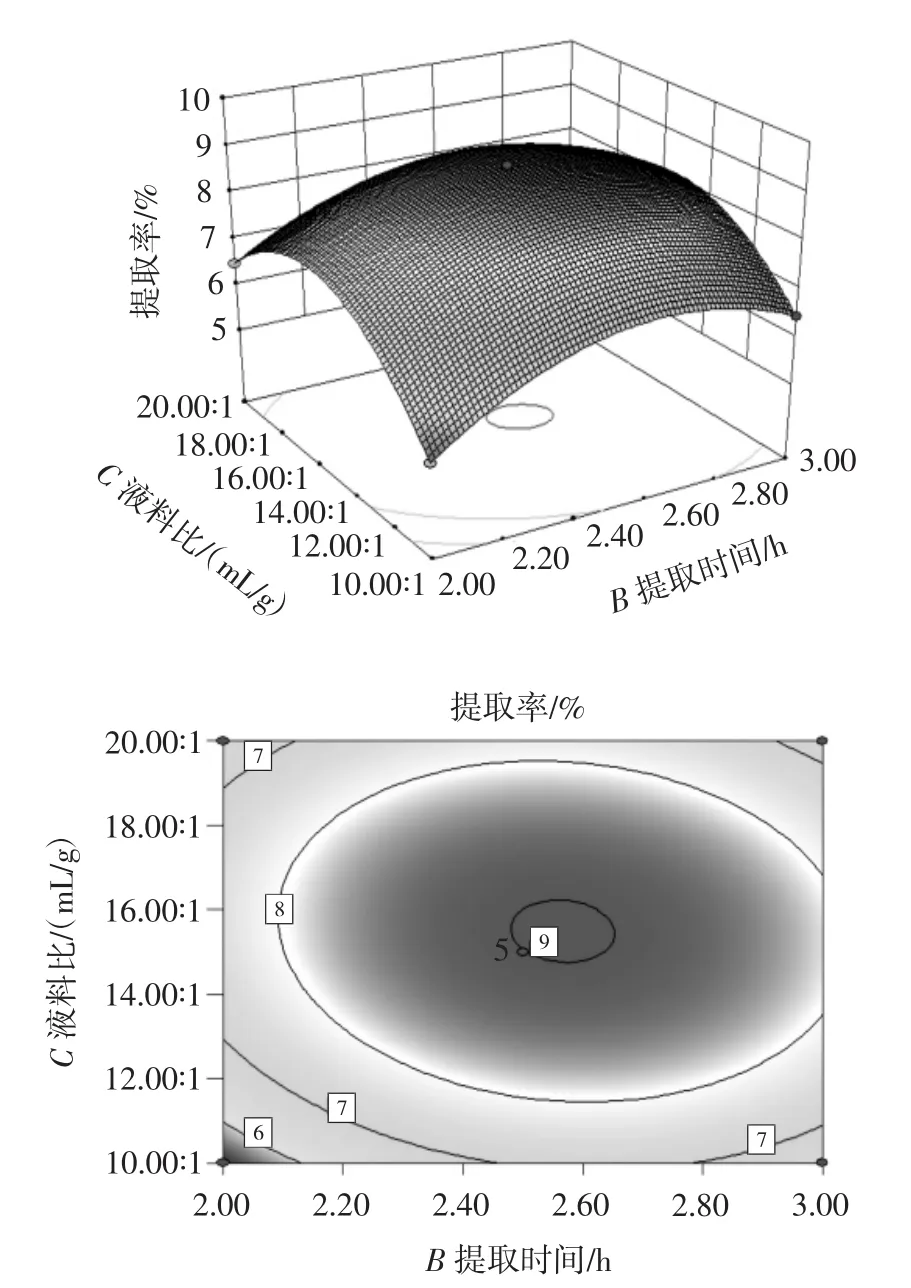
图6 提取时间和液料比两因素对提取率的交互作用Fig.6 Interaction of extraction time and liquid-solid ratio on extraction yield
由图4~图6可知,对提取率影响最大的交互作用是提取温度和提取时间,而提取温度和液料比的影响最小。
通过软件计算后,得到最优的条件为温度80.70℃、时间 2.58h、液料比 15.64∶1(mL/g),预测提取率为9.06%。在实验室实际操作后,确定了最优的条件为提取时间2.5 h、温度 80 ℃和液料比 15∶1(mL/g),测得实际的提取率取平均值为9.03%,相对偏差为0.74%,这表明响应面法优化的提取条件是稳定可行的。
2.3 紫甘薯多糖的分离纯化
洗脱曲线如图7所示,两个峰命名为PSPP-A和PSPP-B。合并收集糖含量最高的PSPP-A,进行后续的纯度鉴定。

图7 紫甘薯多糖的Sepharose 4B洗脱曲线Fig.7 Sepharose 4B elution curve of purple sweet potato polysaccharide
2.4 PSPP-A的纯度及分子量鉴定
多糖分子量标准曲线如图8所示。分子量不同的葡聚糖标准曲线回归方程为Y=-4.310 7X+36.120 0。PSPP-A的液相色谱图如图9所示。
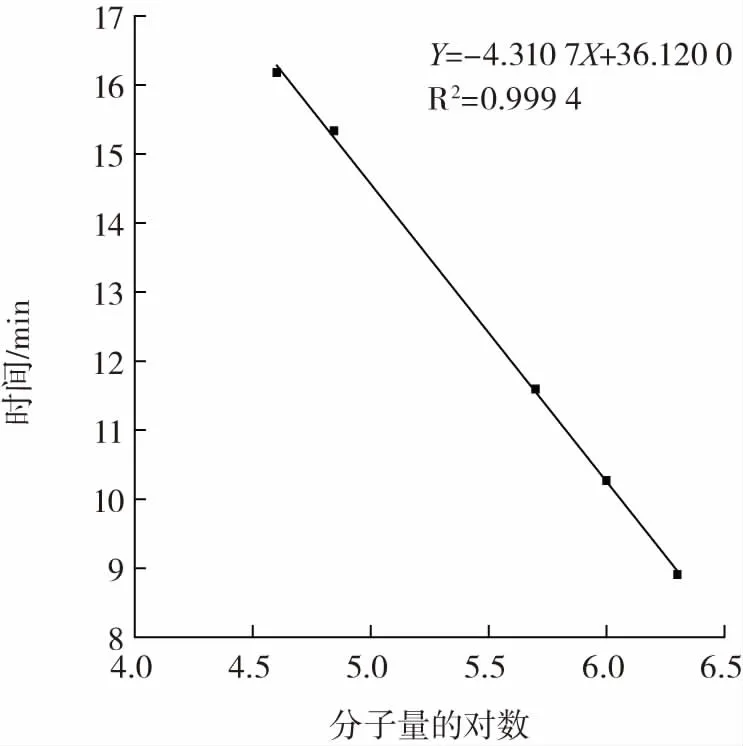
图8 多糖分子量标准曲线Fig.8 Standard curve of molecular weight of polysaccharide
由图9可知,PSPP-A的液相色谱图为单一峰,证明多糖经Sepharose 4B凝胶层析柱纯化后均一性较好。PSPP-A的出峰时间为8.438 min,经过计算,最终得出PSPP-A的分子量为2.63×103kDa。

图9 PSPP-A的高效液相色谱图Fig.9 High performance liquid chromatography of PSPP-A
2.5 PSPP-A的紫外光谱分析
PSPP-A的紫外扫描光谱图见图10。

图10 PSPP-A的紫外扫描光谱图Fig.10 UV scanning spectra of PSPP-A
紫外分光光度计扫描PSPP-A溶液后,260 nm和280 nm处均无明显的吸收峰,说明多糖基本不含核酸、蛋白质类物质[26]。
2.6 PSPP-A的傅里叶红外光谱分析
利用KBr压片法对多糖进行傅里叶红外光谱分析,可以得到PSPP-A的特征官能团和糖环构型等结构信息[27]。PSPP-A的傅里叶变换红外光谱图见图11。
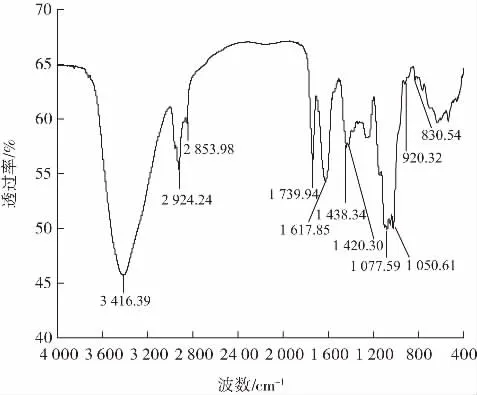
图11 PSPP-A的傅里叶变换红外光谱图Fig.11 Fourier transform infrared spectrumdiagram of PSPP-A
由图11可知,3 416.39 cm-1处的宽吸收峰是—OH伸缩振动,2 924.24 cm-1和2 853.98 cm-1处的吸收峰是C—H的伸缩弯曲振动,这都是多糖的特征峰,因此可以验证PSPP-A是多糖类物质[28]。1 739.94 cm-1处有吸收峰是因为存在酯羧基,说明PSPP-A含有糖醛酸;1 617.85 cm-1处的吸收峰是羧酸盐离子[29]。1 438.34、1 420.30 cm-1处的吸收峰是由于C—H的变角振动[30]。在1 077.59、1 050.61 cm-1处存在C—O—C的弯曲振动,表明存在吡喃糖环[31]。在830.54 cm-1处的吸收峰表明糖结构为α-糖苷键,在920.32 cm-1处的吸收峰归因于 β-糖苷键[32]。
2.7 PSPP-A的单糖组成分析
离子色谱法能使单糖通过金电极表面时发生氧化反应,进而引起电流变化来高效地进行单糖分析[33],不需要进行衍生操作,且每种单糖都会有显著的色谱峰,适合不易提取多糖中的糖类成分分析[34]。PSPP-A经三氟乙酸水解后进行离子色谱分析,结果如图12所示。PSPP-A的单糖组成分析见表4。
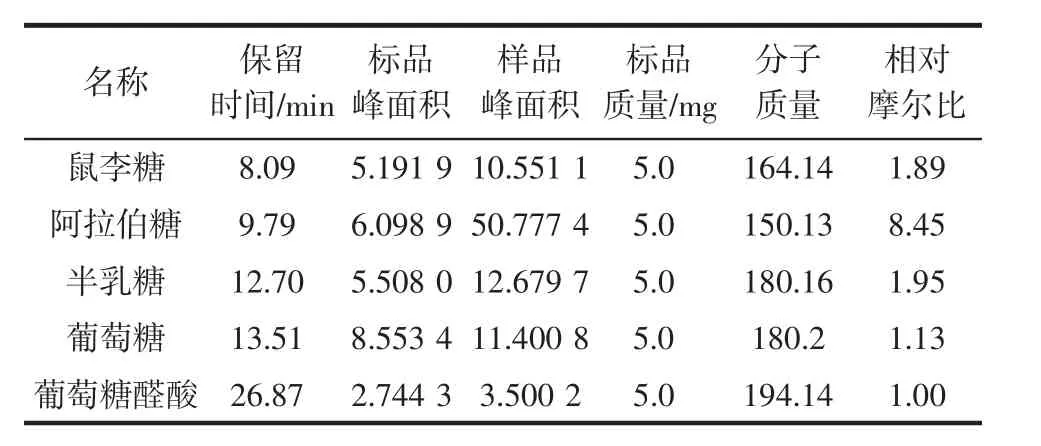
表4 PSPP-A的单糖组成分析Table 4 Monosaccharide composition of PSPP-A

图12 PSPP-A的单糖组成离子色谱图Fig.12 Ion chromatograms for determination of PSPP-A monosaccharide composition
根据图12单糖标准品的出峰顺序和时间判断PSPP-A中的单糖组成为鼠李糖、阿拉伯糖、半乳糖、葡萄糖、葡萄糖醛酸。
由表4可知,PSPP-A单糖(鼠李糖:阿拉伯糖:半乳糖:葡萄糖:葡萄糖醛酸)组成的相对摩尔比为1.89∶8.45∶1.95∶1.13∶1.00。
3 结论
本论文以紫甘薯多糖为研究对象,对其提取率的工艺优化和分离纯化进行了研究,通过进行单因素和响应面试验,得到当提取温度80℃、提取时间2.5 h和液料比15∶1(mL/g)时,紫甘薯多糖提取率最佳,为9.03%。选用Sepharose 4B凝胶分离纯化后得到纯多糖PSPP-A。PSPP-A的相对分子量为2.63×103kDa。通过紫外吸收光谱可知,PSPP-A中不含核酸和蛋白质类物质。PSPP-A的FT-IR图谱存在吡喃糖环、α-糖苷键和β-糖苷键。PSPP-A经单糖组成分析判定后得出,其是由鼠李糖、阿拉伯糖、半乳糖、葡萄糖和葡萄糖醛酸5 种单糖组成,相对摩尔比为 1.89∶8.45∶1.95∶1.13∶1。本研究可为紫甘薯的综合开发和食品功能因子的应用提供参考。
