荆防合剂质量标准提升研究*
2022-12-20庄会芳袁晓梅孙晓兰刘思延范建伟
庄会芳 ,徐 丽 ,袁晓梅 ,孙晓兰 ,刘思延 ,金 凤 ,范建伟 △
(1. 鲁南厚普制药有限公司,山东 临沂 276006; 2. 鲁南制药集团股份有限公司·中药制药共性技术国家重点实验室,山东 临沂 276006)
荆防合剂基于荆防败毒散,经现代中药制药工艺提取浓缩而成,由荆芥、防风、羌活、独活、柴胡、前胡等11 味中药组方,具有发汗解表、散风祛湿功效,主治感冒风寒诸症[1]。荆防合剂在新冠肺炎疫情防控中发挥了积极作用,四川、云南、广东、新疆等地将荆防败毒散及其成方制剂列入防治推荐用药[2]。目前,荆防合剂现行质量标准仍为原卫生部药品标准WS3-B-1377-93,仅规定了制剂的性状、相对密度及其他通则项检验项目,缺乏薄层色谱定性鉴别项和液相色谱含量测定项,难以全面控制该产品的内在质量。且国内荆防合剂的批准文号较多,市售产品质量参差不齐。为确保荆防合剂产品的稳定性和临床用药的安全、有效,本研究中采用薄层色谱法对方中川芎、荆芥、羌活、防风、柴胡、甘草6 味中药进行定性鉴别,采用双波长高效液相色谱法对紫花前胡苷、柚皮苷、新橙皮苷3个指标性成分进行定量检测,为提升荆防合剂的质量标准提供参考。现报道如下。
1 仪器与试药
1.1 仪器
Waters e2695 型高效液相色谱系统(美国Waters 公司),配有光电二极管阵列检测器(PDA)、Empower色谱工作站;TLC Visualizer 2型薄层色谱成像系统(瑞士Camag公司);XS105DU 型电子分析天平(瑞士Mettler-Toledo公司,精度为万分之一);KQ-250DB型数控超声波清洗器(昆山市超声仪器有限公司,功率为450 W,频率为40 kHz);H1650 - W 型台式微量高速离心机(长沙湘仪离心机仪器有限公司);Milli - Q 型超纯水仪(美国Millipore 公司);硅胶G60 薄层板、硅胶G60 F254薄层板(德国Merck公司)。
1.2 试药
胡薄荷酮对照品(批号为111706 - 201907),升麻素苷对照品(批号为111522-201913),柴胡皂苷d对照品(批号为110778-201912),甘草酸铵对照品(批号为110731-201720),紫花前胡苷对照品(批号为111821-201604,含量以99.6%计),柚皮苷对照品(批号为110722 - 201815,含量以91.7%计),新橙皮苷对照品(批号为111857-201804,含量以99.4%计),川芎对照药材(批号为120918 - 201813),均购自中国食品药品检定研究院;荆防合剂(鲁南厚普制药有限公司,规格为每支 10 mL,批号分别为 245210081,245210091,245210101,24521012,24521013,24521014,24521015,24521016,24521017,24521018,依次编号为S1-S10);聚酰胺30-60目(国药集团化学试剂有限公司,批号为F20070411);甲醇、乙腈(色谱纯,美国Tedia 公司);其余试剂均为分析纯,水为自制超纯水。
2 方法与结果
2.1 薄层色谱鉴别
川芎、荆芥:取样品30 mL,置250 mL 烧瓶中,加水70 mL 和沸石数粒,连接挥发油测定器,从测定器上端加水至溢流入烧瓶时为止,再用移液管加入1 mL 乙酸乙酯,连接回流冷凝管[3],加热至沸,并保持微沸30 min,放冷,分取乙酸乙酯层,作为供试品溶液。取胡薄荷酮对照品适量,加甲醇制成每1 mL 含0.5 mg 的溶液,作为对照品溶液。取川芎对照药材0.5 g,加20 mL 甲醇,超声处理30 min,滤过,滤液蒸干,残渣加甲醇1 mL 使溶解,作为对照药材溶液。再按荆防合剂处方工艺分别制备缺川芎和缺荆芥的阴性样品,按供试品溶液制备方法分别制备缺川芎阴性对照品溶液和缺荆芥阴性对照品溶液。按2020 年版《中国药典(四部)》通则0502 薄层色谱法试验,吸取上述5种溶液各10 μL,分别点于同一硅胶G 薄层板上,以石油醚(60~90 ℃)- 乙酸乙酯(9∶1,V/V)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与川芎对照药材溶液色谱相应位置显相同颜色的荧光主斑点,且阴性对照无干扰。详见图1 A。再喷以5%香草醛硫酸溶液,于105 ℃加热至斑点显色清晰,日光下检视。供试品溶液色谱中,在与胡薄荷酮对照品溶液色谱相应位置显相同颜色的斑点,且阴性对照无干扰。详见图1 B。

图1 薄层色谱图Fig.1 TLC chromatograms
羌活:取样品10 mL,用乙酸乙酯振摇提取2 次,每次25 mL,合并乙酸乙酯液,下层水液备用,蒸干乙酸乙酯,残渣加5 mL 水,上聚酰胺柱(3 g,干法上柱,内径10 mm),以50 mL水洗脱,弃去洗脱液,再以40 mL乙酸乙酯洗脱,收集乙酸乙酯洗脱液,蒸干,残渣加甲醇0.5 mL 使溶解,作为供试品溶液。取紫花前胡苷对照品适量,加甲醇制成每1 mL 含0.5 mg 的溶液,作为对照品溶液。再按荆防合剂处方工艺制备缺羌活的阴性样品,按供试品溶液制备方法制备阴性对照品溶液。按2020 年版《中国药典(四部)》通则0502 薄层色谱法试验,吸取上述3 种溶液各5 μL,分别点于同一硅胶G 薄层板上,以三氯甲烷-甲醇(6∶1,V/V)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置显相同颜色的荧光斑点,且阴性对照无干扰。详见图1 C。
防风:取羌活项下下层水液,加80 mL 正丁醇,摇匀,用氨试液洗涤3次,每次30 mL,弃去上清液,取正丁醇液蒸干,残渣加甲醇0.5 mL 使溶解,作为供试品溶液。取升麻素苷对照品,加甲醇制成每1 mL 含1 mg 的溶液,作为对照品溶液。再按荆防合剂处方工艺制备缺防风的阴性样品,按供试品溶液制备方法制备阴性对照品溶液。按2020 年版《中国药典(四部)通则0502 项下薄层色谱法试验,吸取上述供试品溶液、阴性对照品溶液各20 μL 和对照品溶液10 μL,分别点于同一硅胶GF254薄层板上,以三氯甲烷 - 甲醇(5∶1,V/V)为展开剂,展开,取出,晾干,置紫外光灯(254 nm)下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置显相同颜色的斑点,且阴性对照无干扰。详见图1 D。
柴胡:取样品10 mL,加乙酸乙酯振摇提取2 次,每次20 mL,合并乙酸乙酯液,用氨试液洗涤2 次,每次30 mL,弃去上清液,取乙酸乙酯液蒸干,残渣加甲醇0.5 mL 使溶解,再加稀盐酸2 滴,摇匀,作为供试品溶液。取柴胡皂苷d 对照品适量,加甲醇制成每1 mL 含1 mg 的溶液,再加稀盐酸2 滴,摇匀,作为对照品溶液。再按荆防合剂的处方工艺制备缺柴胡的阴性样品,按供试品溶液制备方法制备阴性对照品溶液。按2020 年版《中国药典(四部)》通则0502 薄层色谱法试验,吸取上述3种溶液各10 μL,分别点于同一硅胶G 薄层板上,以三氯甲烷 - 甲醇 - 水(13∶7∶2,V/V/V)10 ℃以下静置的下层溶液为展开剂[4-5],展开,取出,晾干,喷以5%对二甲氨基苯甲醛的10%硫酸乙醇溶液,于60 ℃加热至斑点显色清晰,日光下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置显相同颜色的斑点,且阴性对照无干扰。详见图1 E。
甘草:取样品10 mL,加5 mL稀盐酸,超声处理10 min,离心,弃去上清液,沉淀用10 mL 0.5%碳酸氢铵溶液溶解,加水饱和正丁醇振摇提取2 次,每次20 mL,弃去正丁醇液,下层水液加冰醋酸3 mL,摇匀,再加水饱和的正丁醇溶液振摇提取2 次,每次20 mL,合并正丁醇液,蒸干,残渣用0.5 mL 甲醇溶解,作为供试品溶液。取甘草酸铵对照品,加甲醇制成每1 mL 含1 mg 的溶液,作为对照品溶液。再按荆防合剂的处方工艺制备缺甘草的阴性样品,按供试品溶液制备方法制备阴性对照品溶液。按2020 年版《中国药典(四部)》通则0502 薄层色谱法试验,吸取上述3 种溶液各5 μL,分别点于同一硅胶GF254薄层板上,以乙酸乙酯 - 甲酸 - 冰醋酸 - 水(15∶1∶1∶2,V/V/V/V)为展开剂,展开,取出,晾干,置紫外光灯(254 nm)下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置显相同颜色的斑点,且阴性对照无干扰。详见图1 F。
2.2 紫花前胡苷、柚皮苷、新橙皮苷含量测定
2.2.1 色谱条件与系统适用性试验
参照2020 年版《中国药典(四部)通则0512 高效液相色谱法和参考文献[6 - 9]开展预试验,确定如下色谱条件。色谱柱:Agilent Zorbax SB C18柱(150 mm×4.6 mm,3.5 μm);流动相:乙腈(A)-0.1%磷酸水溶液(B),梯度洗脱(0~35 min 时 16%A,35~36 min 时 16%A →90%A,36~44 min 时90%A);流速:1.0 mL/ min;检测波长:336 nm(紫花前胡苷),283 nm(柚皮苷和新橙皮苷);柱温:25 ℃;进样量:5 μL。在此色谱条件下,紫花前胡苷、柚皮苷、新橙皮苷的理论板数均大于3 000,分离度均大于1.5。
2.2.2 溶液制备
精密吸取荆防合剂2 mL,置50 mL 容量瓶中,用30%甲醇稀释并定容,摇匀,以0.45 μm微孔滤膜滤过,取续滤液,即得供试品溶液。分别取紫花前胡苷对照品、柚皮苷对照品、新橙皮苷对照品16.97,28.59,21.84 mg,精密称定,分别置50 mL容量瓶中,以50%甲醇溶解并定容,摇匀,即得各单一成分对照品贮备液。分别精密吸取各单一成分对照品贮备液4,3,3 mL,置同一25 mL容量瓶中,用50%甲醇稀释并定容,摇匀,即得每1 mL分别含紫花前胡苷、柚皮苷、新橙皮苷54.08,62.92,52.10 μg 的混合对照品溶液。根据荆防合剂的处方工艺,分别制备缺羌活的阴性样品和缺枳壳的阴性样品,并按供试品溶液制备方法制备缺羌活阴性对照品溶液和缺枳壳阴性对照品溶液。
2.2.3 方法学考察
专属性试验:分别精密吸取2.2.2 项下供试品溶液、混合对照品溶液和阴性对照品溶液各5 μL,按2.2.1项下色谱条件进样测定,记录色谱图。结果显示,相应检测波长下,供试品溶液、对照品溶液色谱图中,紫花前胡苷、柚皮苷、新橙皮苷在各自的保留时间处均有吸收峰,且峰形对称;相应阴性对照品溶液色谱图中,在紫花前胡苷和柚皮苷、新橙皮苷相应保留时间处均无吸收峰,表明方法专属性强,阴性对照无干扰。详见图2。
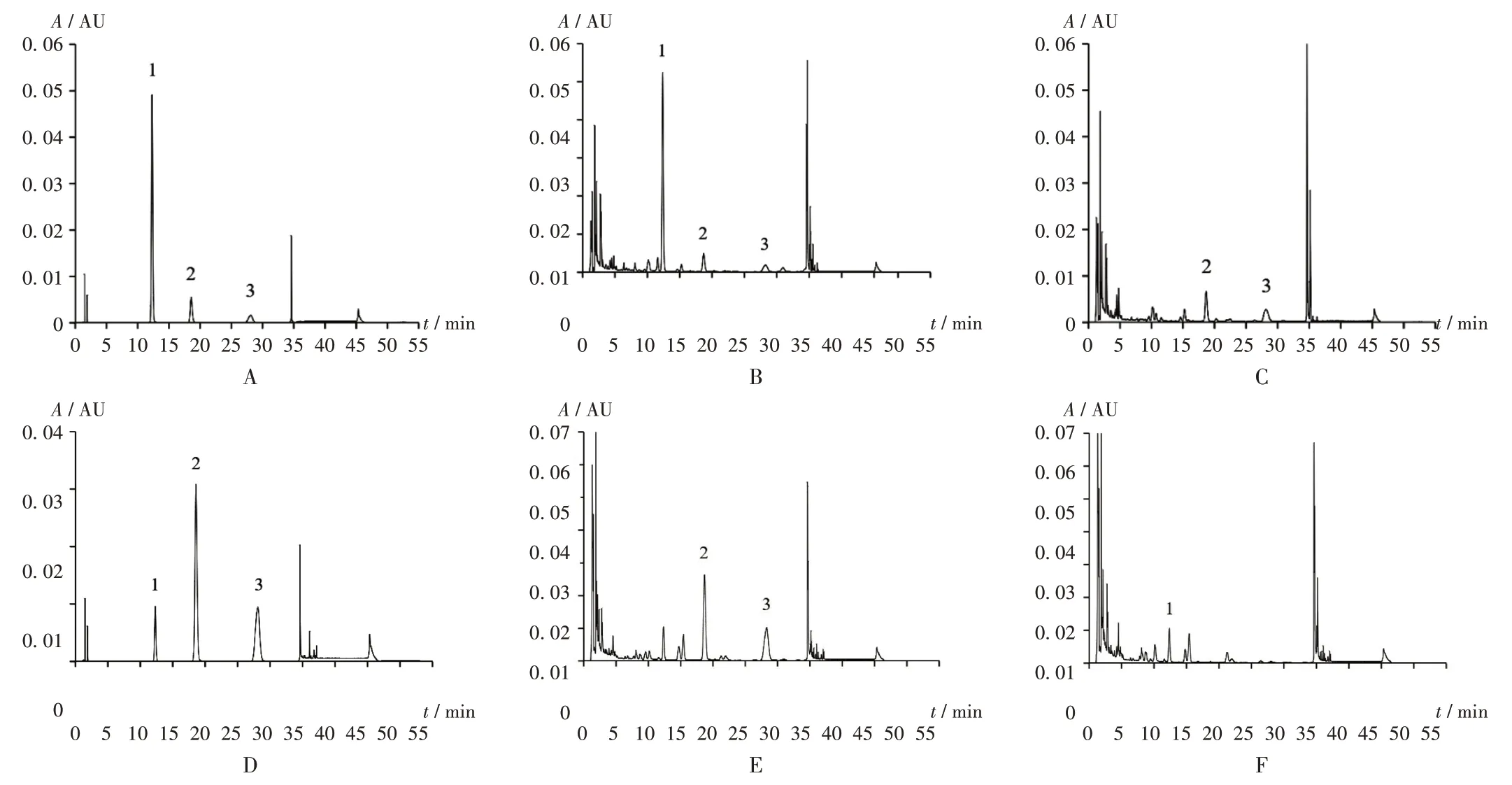
图2 高效液相色谱图Fig.2 HPLC chromatograms
线性关系考察:精密吸取2.2.2项下单一成分对照品贮备液各适量,依次置相同容量的不同容量瓶中,分别用50%甲醇制备系列质量浓度的混合对照品溶液。其中,紫花前胡苷的质量浓度分别为10.14,20.28,33.80,50.70,67.60,135.20 μg/mL,柚皮苷的质量浓度分别为10.49,20.97,52.43,78.64,104.86,157.29 μg/mL,新橙皮苷的质量浓度分别为8.68,17.37,34.74,43.42,65.13,86.84 μg/mL。精密吸取上述系列混合对照品溶液各适量,按2.2.1 项下色谱条件进样测定,以峰面积(Y)为纵坐标、进样质量浓度(X,μg/mL)为横坐标进行线性回归,得紫花前胡苷、柚皮苷、新橙皮苷的回归方程分别为Y紫=1.319×104X紫-1.773×10(4r=0.999 8),Y柚=9.078 × 103X柚-1.365 × 104(r=0.999 4),Y新=9.719×103X新+8.804×10(3r=0.999 5)。结果表明,紫花前胡苷、柚皮苷、新橙皮苷的质量浓度分别在 10.14~135.20 μg/ mL,10.49~157.29 μg/ mL,8.68~86.84 μg/mL范围内与相应峰面积线性关系良好。
精密度试验:精密吸取2.2.2 项下混合对照品溶液各适量,按2.2.1 项下色谱条件连续进样测定6 次,记录峰面积。结果紫花前胡苷、柚皮苷、新橙皮苷峰面积的RSD分别为0.30%,0.31%,0.47%(n=6),表明仪器精密度良好。
稳定性试验:取同一批(编号为S1)供试品溶液,分别于制备后0,2,4,8,12,24 h时按2.2.1项下色谱条件进样测定,记录峰面积。结果紫花前胡苷、柚皮苷、新橙皮苷峰面积的RSD分别为0.50%,0.55%,0.55%(n=6),表明供试品溶液在24 h内稳定性良好。
重复性试验:取样品(编号为S1)适量,精密称定,按2.2.2 项下方法平行制备供试品溶液6 份,按2.2.1项下色谱条件进样测定,计算得紫花前胡苷、柚皮苷、新橙皮苷的平均含量分别为1.48,1.73,1.21 mg/g,结果的RSD分别为0.71%,0.86%,0.85%(n=6),表明方法重复性良好。
加样回收试验:依次取紫花前胡苷对照品、柚皮苷对照品、新橙皮苷对照品14.87,18.87,12.13 mg,精密称定,置同一50 mL容量瓶中,用30%甲醇溶解并定容,摇匀,即得混合对照品贮备液(紫花前胡苷、柚皮苷、新橙皮苷的质量浓度依次为296.21,346.08,241.14 μg/mL);精密吸取已知含量的样品(编号为S1)1 mL,共6份,分别置50 mL 容量瓶中,各精密加入上述混合对照品贮备液5 mL,按2.2.2项下方法制备供试品溶液,按2.2.1项下色谱条件进样测定,并计算回收率。结果见表1。

表1 加样回收试验结果(n=6)Tab.1 Results of the recovery test(n=6)
2.2.4 样品含量测定
取10 批样品(编号为S1-S10),按2.2.2 项下方法制备供试品溶液,各平行3 份,按2.2.1 项下色谱条件进样测定,以外标法计算各成分的含量,结果见表2。

表2 样品含量测定结果(n=3)Tab.2 Results of content determination of nodakenin,naringin and neohesperidin in the samples(n = 3)
3 讨论
3.1 薄层色谱鉴别
本研究在川芎、荆芥的鉴别中,采用蒸馏油法制备供试品溶液,并用同一展开剂分别实现了川芎(显色前)和荆芥(显色后)的定性鉴别;在羌活、防风的鉴别中,采用“步进式”供试品处理方法,简化了操作,提高了检测效率;在柴胡的鉴别中,通过滴加稀盐酸使柴胡皂苷能有效转化为皂苷元,结果显色斑点更清晰、稳定,特征性更强;在甘草的鉴别中,结合大量预试验结果,通过调节溶液酸碱性实现对甘草中甘草酸铵的有效鉴别。
在展开系统的选择方面,针对羌活的鉴别,因紫花前胡苷为羌活主要活性成分[10],故以紫花前胡苷为对照,确定三氯甲烷 -甲醇(6∶1,V/V)为展开剂;针对防风的鉴别,参考了三氯甲烷 - 甲醇(4∶1,V/V)[11]156、二氯甲烷 - 甲醇 - 甲酸(16∶4∶1,V/V/V)[12]等不同展开系统,最终确定以三氯甲烷-甲醇(5∶1,V/V)为展开剂。
3.2 含量测定
分别考察了乙腈-0.2%甲酸水溶液、乙腈- 0.1%磷酸水溶液和甲醇- 0.2%甲酸水溶液、甲醇- 0.1%磷酸水溶液4 组流动相系统,结果在乙腈- 0.1%磷酸水溶液系统下,供试品溶液色谱图基线平稳;相应检测波长下,紫花前胡苷、柚皮苷、新橙皮苷色谱峰的对称性和分离度均较好,故选用乙腈-0.1%磷酸水溶液为流动相,以梯度洗脱模式进行分析。由于羌活中紫花前胡苷在336 nm 波长处有最大紫外吸收,而枳壳中的柚皮苷、新橙皮苷在283 nm 波长处有最大紫外吸收,故在同一液相色谱条件下,采用双波长法对这3个指标性成分进行定量分析,且分离效果良好,阴性对照无干扰。此外,同时测定这3 个指标性成分的含量,较单一成分检测更科学、合理。
本研究中10 批荆防合剂生产投料所用羌活药材中,紫花前胡苷含量介于18.59~26.78 mg/ g,平均22.68 mg/g,其成分转移率以80%计,暂定羌活中紫花前胡苷含量的最低限度为18.14 mg/g。所用枳壳药材中,柚皮苷含量介于40.06~52.75 mg/g,平均46.40 mg/g;新橙皮苷含量介于31.22~39.53 mg/g,平均35.38 mg/g,二者均高于2020年版《中国药典(一部)》中枳壳项下柚皮苷含量的最低限度40.0 mg/g和新橙皮苷含量的最低限度30.0 mg/g[11]257。结合 2.2.4 项下含量测定结果,计算得紫花前胡苷、柚皮苷、新橙皮苷3 个指标性成分含量的转移率分别介于60.05%~86.69%(平均73.37%)、38.43%~50.87%(平均44.65%)、35.26%~37.54%(平均36.40%)。按平均转移率的80%计算,则荆防合剂中紫花前胡苷、柚皮苷、新橙皮苷的含量下限分别为1.07,1.39,0.85 mg/mL。故暂定本品每1 mL 含紫花前胡苷不得少于1.0 mg,含柚皮苷不得少于1.3 mg,含新橙皮苷不得少于0.8 mg。
3.3 方法评价
本研究中建立的方法操作简单、结果可靠、专属性强、重复性好,可用于荆防合剂的质量控制。
