基于SNP共表达网络肝癌分子分型及预后分析
2022-12-17张思嘉
张思嘉,蔡 挺,张 顺
(中国科学院大学 宁波华美医院医学实验部,浙江 宁波 315100)
肝癌是目前我国第4位常见恶性肿瘤及第2位肿瘤致死病因,全世界每年近一半的新发和死亡病例发生在中国[1]。目前原发性肝癌的治疗方法虽然很多,但肿瘤治疗的预后效果仍不理想:HCC首选方法是手术切除,但由于其起病隐蔽,确诊时患者多已中晚期,失去手术切除机会[2]。而传统的放化疗等手段预后较差,不良反应较多,这使目前我国HCC的临床治疗充满挑战,急需新的治疗策略来改善肝癌患者的生存质量。
肝癌发生是多因素、多步骤和受多种机制调控的复杂过程,具有高度异质性。目前针对肝癌的大体分型和组织病理学分型是制定临床治疗方案,预测判断患者的预后与转归的重要依据。但实践表明许多具有相同类型和相同分期的肝癌的患者应用相似的临床治疗手段,其预后的差别很大,这与肝癌极其复杂的肿瘤异质性密切相关。因此迫切需要新的分期分型指标以助力肝癌精准诊断与治疗,以提高患者生存率。随着二代测序(Next-generation Sequencing,NGS)技术,基因组、转录组、单细胞测序等多组学的发展,这些技术逐渐被应用于肿瘤的发病机制的研究以及分子分型的判断中,并为诊治手段及预后分析提供重要信息[3-4]。而基于肿瘤分子异质性的个体化精准诊疗已成为未来恶性肿瘤诊疗的发展方向:该诊疗方式将传统的肝癌临床病理分型和分期与肝癌的分子表型相结合,对肝癌患者进行更加细致的亚群划分,并在这一基础上将手术、放疗、化疗、免疫治疗、分子靶向治疗等手段,按照患者的个体化特征定制精准治疗方案,从而大幅度提高治疗手段的针对性和治疗效果[5-6]。
单核苷酸多态性(SNPs)是指基因组DNA序列中由于单个核苷酸替换而引起的多态性,是最普遍的遗传变异形式。绝大多数SNP并不影响蛋白序列,而是通过对基因表达的调控对生物个体产生影响[7]。越来越多的临床研究证实,通过监测肿瘤患者中生物标志物的SNP突变、mRNA及蛋白的表达水平的异常变化,来评价预后并指导临床个体化治疗,可提高疗效果,减轻不良反应,促进医疗资源的合理利用[8-10]。本研究利用肝癌患者的转录组和SNP突变数据,对样本进行分子分型并分析不同分型间的生物学差异,有利于进一步认识肝癌发生和进展的过程,并对肝癌的临床诊断和治疗选择以及预后预测具有一定的参考价值。
1 材料和方法
1.1 数据来源
本研究涉及的359例肝细胞癌患者的mRNA表达数据、单核苷酸突变数据及临床数据下载于TCGA在线公共数据库(https://portal.gdc.cancer.gov)。
1.2 分析方法
1.2.1 SNP突变与mRNA表达谱联合分析
单核苷酸突变(SNP)可以通过影响基因编码和剪接影响基因表达[11]。本研究通过对肝癌患者SNP突变数据和mRNA表达谱的联合分析,筛选出因单核苷酸突变引起表达水平变化的差异基因,具体实施方式如下:首先根据SNP突变数据中基因是否发生突变,将肝癌患者划分为野生型和突变型两组,再通过wilcoxon秩和检验比较野生型和突变型肝癌患者中的表达水平,其中表达水平存在存在显著差异的基因作为单核苷酸突变差异表达基因(取P<0.01为差异具有统计学意义)。最后运用KEGG数据库通路富集和基因功能注释的信息,对单核苷酸突变表达差异基因进行功能聚类分析和代谢途径分析,并选取与癌症相关的单核苷酸突变差异表达基因进行后续分析。
1.2.2 蛋白互作网络的构建与枢纽基因的筛选
将与癌症相关的单核苷酸突变差异表达基因导入STRING数据库(https://www.string-db.org),得到相互作用的网络节点文件,再通过Cytoscape软件对蛋白-蛋白相互作用网络PPI(Protein-protein Interaction)进行可视化处理,同时用插件CytoHubba对PPI网络进行模块分析,选取出10个连接度最高的Hub 基因作为肝癌分子分型的潜在特征和重要依据。
1.2.3 肝癌患者分子分型与预后分析
非负矩阵分解(Nonnegative Matrix Factorization,NMF)属于双向聚类,具有良好的可解释性和数值结果,该方法已广泛用于基因表达谱数据的癌症分类[12-14]。本研究利用R语言中的“Consensus ClusterPlus”分析包,根据肝癌患者10个hub基因的mRNA表达量构建NMF分子分型模型,聚类数k值取2~8,根据聚类效果选取具有较好聚类稳定性的k值用于分子分型的划分,并进一步将样本的基因表达数据和临床预后信息整合,去除生存时间小于30天的样本,再利用R软件中的"Survival"包,对不同分子分型的肝癌患者进行Kaplan-Meier 生存分析及log-rank检验,判断不同分子分型患者的生存预后是否具有显著性差异。
1.2.4 不同分子分型肝癌患者转录组特征对比
首先采用R软件的DESeq2软件包,对不同分子分型患者的基因转录组数据进行差异表达分析,差异基因的筛选标准为:算法矫正假发现率(FDR)<0.05,差异倍数(Fold change)> 1。再结合样本的分子分型分组,针对差异表达基因利用R软件的WGCNA 软件包进行加权共表达网络分析,具体步骤如下:1)构建基因共表达相似性矩阵,确定软阈值后再将相似性矩阵转换为邻接矩阵。2)将邻接矩阵转换成拓扑矩阵,并采用拓扑覆盖法对差异基因进行层次聚类,按照混合动态剪切树的方法确定基因模块,同时绘制树状图并对基因模块进行可视化,其中每个模块的最小基因数目为30。3)将基因模块与分子分型进行关联,寻找与2种分子分型最显著相关的基因模块[15]。并对与分子分型相关的基因模块中的基因利用R软件“Cluster Profiler”包进行KEGG富集分析,预测与2种肝癌分子分型相关的功能和信号通路。
2 结 果
2.1 单核苷酸突变差异表达基因的筛选与蛋白互作网络的构建
通过对癌症基因组图谱(TCGA)数据库中获得肝癌患者SNP突变数据的统计分析,共发现1 932种突变率大于5%的突变基因。根据基因是否发生突变,并通过对突变基因在突变型和野生型两组患者mRNA表达谱的差异分析,确定561种在突变前后的表达水平发生显著性变化的基因作为单核苷酸突变差异表达基因,进一步通过Gene ontology (GO)富集分析进行筛选,最终共得到64种与癌症发生发展相关的单核苷酸突变差异表达基因。将上述基因输入STRING网站,导入Cytoscape软件得到可视化PPI网络图,显示有58个节点和220个边(见图1)。运用CytoHubba插件,计算每个基因得连接度,得分最高的前10个基因作为hub基因(见表1)。其中大多数核心基因的SNP突变与多种肿瘤有关,比如单核苷酸突变引起的KRAS激活可导致多种恶性肿瘤,包括肺腺癌、粘液腺瘤、胰腺导管癌和大肠癌;SMAD4作为一种肿瘤抑制基因,其突变失活引起的TGF-β信号转导紊乱导致对肿瘤细胞生长抑制作用的逃逸[16]。TSC2是常染色体基因,在体内作为抑癌基因广泛表达[17]。STK11突变导致的表达缺失已被证明是肠道息肉综合征(PJS)重要致病原因并与散发性的结直肠癌直接相关[18]。TP53基因(编码p53蛋白)作为一个重要的抑瘤癌基因,通过调控一系列信号转导通路广泛参与了多种恶性肿瘤的发生发展,TP53突变在肝癌中有着较为明显的特征[19]。CTNNB1是Wnt通路的关键成员,参与细胞间黏附以及细胞间信号传递,与肿瘤的形成和浸润,转移密不可分[20]。mTOR控制蛋白质合成、细胞生长和增殖,是PI3K—Akt—mTOR信号通路的核心基因[21]。对10种hub基因进行进一步生存分析,发现共有7种hub基因与肝癌患者生存预后显著相关。其中高表达的CDKN2A、KARS是预后的危险因素,低表达的FOXO1、CDKN1A、MTOR、STK11、TP53是预后的危险因素。由此可见Hub基因的表达异常与肝癌患者的预后关系密切,这些Hub基因有望成为肝癌患者早期诊断、治疗及预后判断的重要靶点。

表1 蛋白质互作网络中连接度排名前 10 核心基因及生存分析结果4Table 1 Ten hub genes in PPI networkand survival analysis(top ten in connectivity)
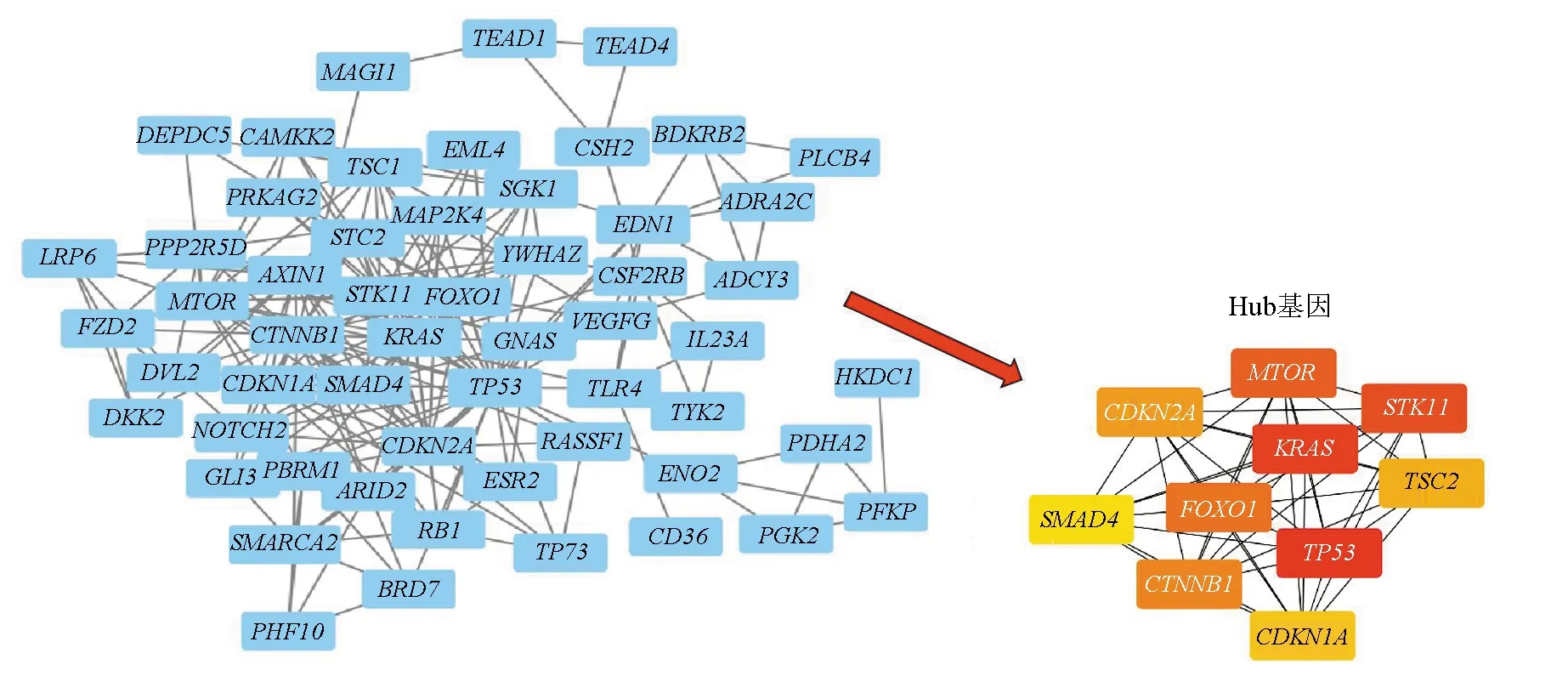
图1 突变后表达显著差异的癌症相关基因构建的PPI网络和hub基因模块Fig.1 PPI network and hub gene module of cancer-associated genes which are significant differentially expressed after mutation
2.2 肝癌分子分型的建立与预后分析
基于2.1筛选出的10个hub基因的表达量,运用非负矩阵因子分解(NMF)算法对肝癌患者进行分子分型。综合判断聚类稳定性,发现当k=2时,即肝癌样本分为2种分子分型时,模型的稳定性较好且样本分布均匀。生存曲线(见图2)以及log-rank检验结果显示2种肝癌分子分型患者的预后情况具有显著性差异,其中cluster1的5年生存率显著低于cluster2(P=0.039)。因此本研究按照2种肝癌分子分型患者生存率的高低将cluster1作为高危组,cluster2作为低危组。由于NMF算法无法直接对因子的贡献度进行统计,为进一步寻找对肝癌分子分型的影响最为显著的Hub基因,本文以Heatmap的形式对比10个Hub基因在高危组(Cluster1)和低危组(Cluster2)的表达趋势,其中基因表达趋势与样本分子分型相关性最为明显的Hub基因将作为后续研究重点。突变基因CDKN2A和FOXO1表达量与样本分子分型的分布最具有明显的趋势性(见图3)。具体表现为CDKN2A在高危组(Cluster1)中的表达量明显高于低危组(Cluster2),FOXO1在低危组(Cluster2)中的表达量明显高于高危组(Cluster1)。并且进一步研究发现,在不考虑分子分型对全部肝细胞癌患者进行生存分析,CDKN2A基因高表达和FOXO1基因低表达的患者生存时间显著降低(见图4)。这与高、低危组两种分子分型组间的生存率高低差异相一致。由此可以初步推测:由突变导致的CDKN2A与FOXO1表达量变化对肝癌样本分子分型贡献较大,其中CDKN2A为预后不良基因(Unfavorable gene),FOXO1为预后良好基因(Favorable gene),因此以CDKN2A、FOXO1突变为基础的分子分型方法具有较大的研究潜力。

图2 2个肝细胞癌分子分型的生存曲线Fig.2 Survival analysis of two HCC molecular typing
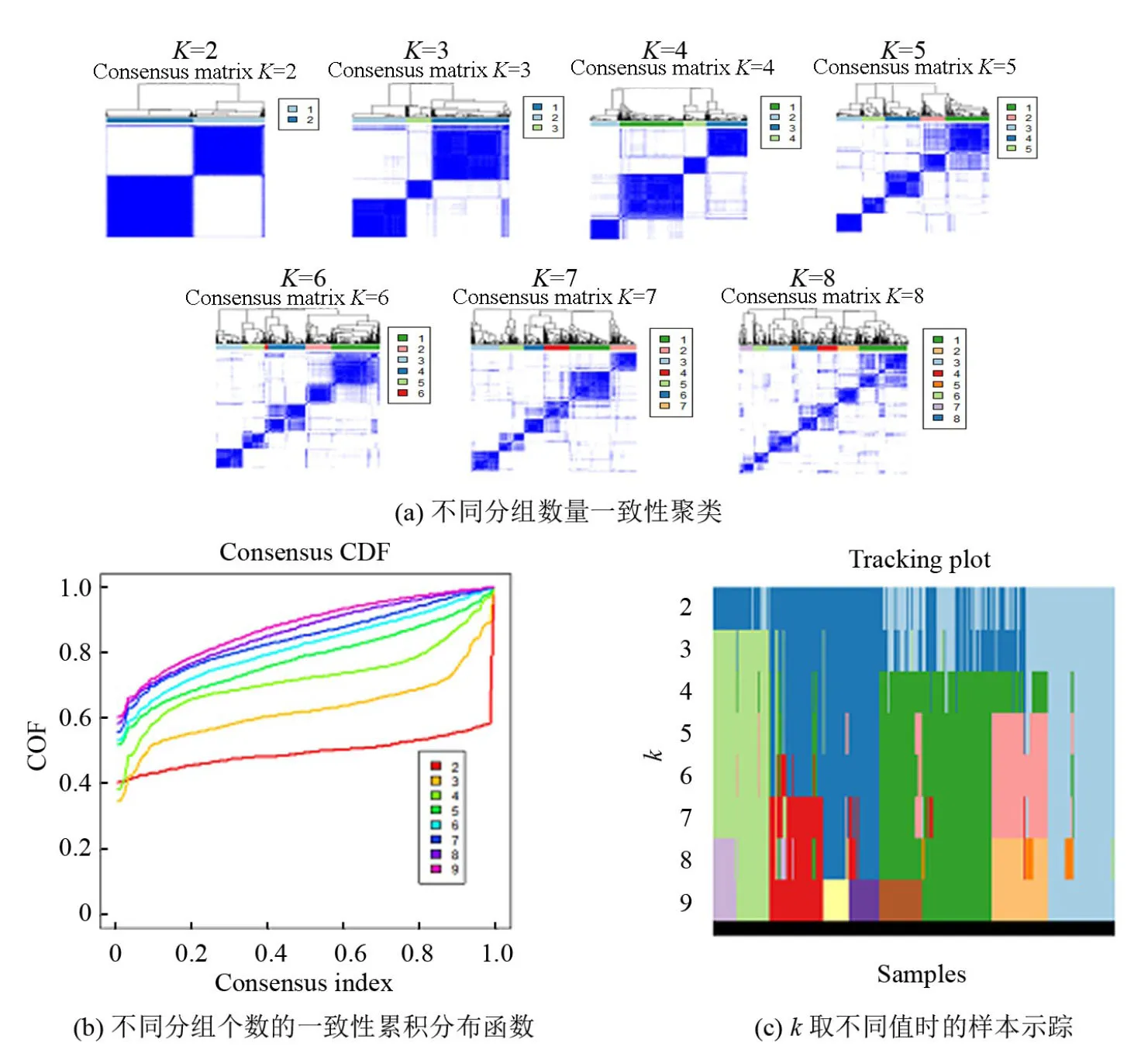
图3 基于NMF模型构建肝细胞癌分子分型Fig.3 Molecular classification of HCC based on NMF model

图4 10种hub基因在不同肝细胞癌分子分型表达水平Fig.4 Expression level of ten hub genes in different molecular typing of HCC
由以上分析表明,基于10种hub基因表达水平预测的分子分型有望成为潜在肝癌的有效预后指标。本研究通过进一步构建肝癌1~5年死亡概率预测列线图,比较肝癌分子分型与其它临床预后变量的关系,从而为肝癌患者的预后情况提供个体化预测。其中临床预后变量均为分类变量,包括肝癌的TNM分期变量、性别、年龄分层,以及治疗方法等,并按照临床预后变量的重要性由下至上进行排列,每一个预后变量均包括若干个变量取值,每一个变量取值对应分值标尺上的一个分值,总分值标尺和患者死亡概率变量均为连续性变量。患者肝癌分子分型在预测模型中的重要程度仅次于T分期,且优于M、N分期以及性别年龄分层(见图5)。该列线图模型的预测准确性较高,1年生存率的AUC为0.762,3年生存率的AUC为0.749,5年生存率的AUC为0.732。由此可以看出基于10中hub基因构建的肝癌分子分型方法在对肝癌患者的病情评估和预后判断具有一定的临床指导意义。

图5 包含年龄(age)、性别(gender)、肿瘤分期(TMN)、治疗方法(treatment type)和分子分型(cluster)等预测因素的肝癌患者1年、3年、5年死亡率的列线图预测模型Fig.5 Nomogram including age, gender,TMN stage, treatment type,and molecular cluster for 1-,3-, and 5-year overall death rate in patients with HCC
为进一步验证分子分型方法的可靠性和稳定性,基于GEO(Gene Expression Omnibus)数据库的GSE76427数据集测序数据及临床信息,采用相同分子分型方法对该数据集中的115例肝癌患者进行预测,并对预测结果中的不同分型患者进行生存分析,同时针对分子分型方法中的关键基因CDKN2A和FOXO的表达水平进行差异分析。如图5所示验证集GSE76427与TCGA数据库的结论相似:生存分析结果均提示cluster2(低危险组)的预后显著优于cluster1(高危险组),且关键基因CDKN2A和FOXO1的表达水平也分别符合在cluster2(低危组)和cluster1(高危组)高表达。
2.4 WGCNA 构建基因共表达模块
为进一步分析影响2种分子分型的肝癌患者分子分型生存率差异的原因,探究高、低危组患者的分子遗传调控机制的不同。本研究首先在转录组水平上,对高危组(Cluster1)和低危组(Cluster2)肝癌患者的转录组表达谱进行差异分析,共筛选出显著差异表达的mRNA共186个(差异倍数变化值大于2或小于0.50且P<0.05),其中高危组(Cluster1)相较于低危组(Cluster2)共有120个下调基因及17个上调基因(见图5),10种hub基因表达在高危组,低危组中均具有统计学差异,其中CDKN1A和FOXO1在高危组的表达显著低于低危组,KARS和CDKN2A在高危组的表达显著高于低危组,这些hub基因的在高、低危组的表达趋势与生存分析结果相一致(见表1)。其次利用加权基因共表达网络分析算法(WGCNA),针对差异表达基因构建共表达模块,并分别对模块进行富集分析,重点挖掘和癌症发生发展相关的通路,并展开更深层次的探索。
通过WGCNA分析共鉴定出5个共表达模块,由于灰色模块(MEgrey)由没有共表达的游离基因组成,因此最终确定的有效模块数目为4个(见图6),每个模块包含基因数目大于30。为确定与分子分型显著相关的特异性基因模块,将基因模块与肝癌分子分型进行关联分析,并计算基因模块与高、低危两种分型的相关性系数和P值。结果显示(见图7) MEblue模块与高危组(Cluster1)具有显著正相关性(r=0.34,P=3×10-11),而MEbrown、MEgreen模块与高危组(Cluster1)具有显著的负相关性(r=-0.4,P=2×10-15)。此外不同基因模块具有特异性的代谢通路及生物学过程,其中MEblue模块基因在TECM-受体相互作用、PI3K-Akt信号通路、黏附信号等3个通路上显著富集,而MEgreen、MEyellow模块则分别与胰腺分泌、糖胺聚糖生物合成-硫酸乙酰肝素/肝素等通路密切相关,MEbrown模块中的基因则主要参与细胞周期的调控(见图8)。结合WGCNA分析和KEGG富集结果,推测高危组富集多种与肿瘤发生发展相关的基因与通路,其中包括ECM-受体相互作用,黏附信号通路,以及与肿瘤细胞增殖相关的PI3K-Akt信号通路,而低危组中主要是与细胞周期的改变相关。类似结果在曹颖颖等人在对比高危组和中低危病胃癌患者分析也有所提及(见图9)[22]。

图6 GSE76427数据集对肝癌分子分型方法的验证Fig.6 Validation for molecular classification method of HCC by GSE76427 dataset

表2 主要基因模块的KEGG通路富集结果Table 2 KEGG analysis of genes in major gene modules
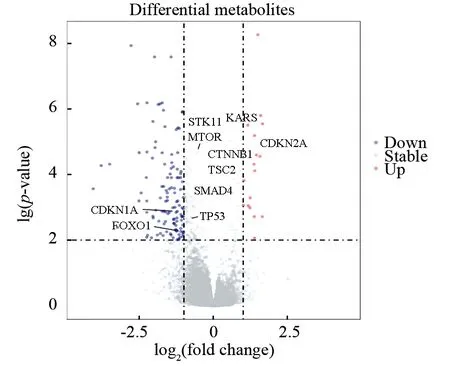
图7 肝癌分子分型差异表达基因的火山图Fig.7 Volcanic map of differentially expressed genes ofdifferent molecular typing of HCC注:左侧蓝点为高危组表达水平显著高于低危组的基因,右侧红点为高危组表达水平显著低于低危组的基因;10种Hub基因具有文字标注.

图8 联合加权共表达网络:基因层次聚类树及基因模块Fig.8 WGCNA: clustering dendrogram of genes with assigned modules
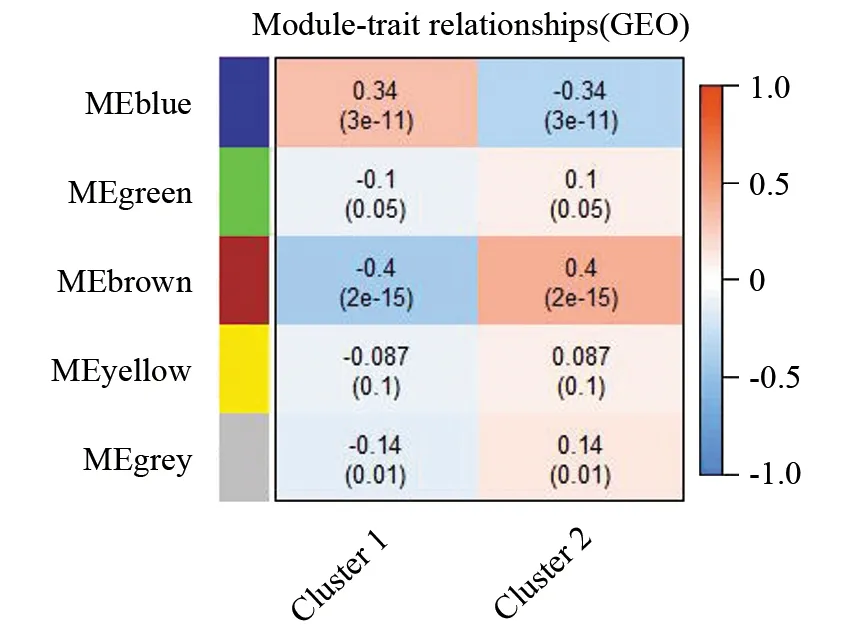
图9 基因模块与肝细胞癌不同分子分型的相关分析Fig.9 Associations between gene modules and different molecular typing of HCC
3 讨 论
通过SNP和mRNA表达谱关联分析及分子分型模型的构建,本研究基于10种核心基因的表达水平将患者分为高危组和低危组两类,二者的生存率存在显著差异。其中核心基因CDKN2A和FOXO1在肝癌分子分型中起到决定性作用,因此我们推测由突变导致的CDKN2A高表达和FOXO1低表达可能与肝癌患者的不良预后密切相关。CDKN2A即细胞周期依赖性激酶抑制基因(Cyclin-dependentkinase inhibitor) ,是一种直接参与细胞周期调控的抑癌基因,由Kamb等于1994年首次报道[23],并以其在肿瘤细胞中的高突变率和抑癌作用而在肿瘤遗传学和肿瘤分子生物学的研究中受到了广泛关注。研究表明CDKN2A的缺失、突变和甲基化可导致其编码两种细胞周期抑制蛋白p16INK4a和p14ARF异常,其中p16INK4a不能与CDKN2A和CDK6结合,使pRb蛋白磷酸化和转录调节因子E2F释放,诱导G1期停滞,有助于肿瘤的发生[24],而p14ARF功能的分子机制比较复杂,可能是通过P14AFP-MDM2-p53途径起作用[25-26],这两条途径的异常普遍存在于各种肿瘤。但本研究基于TCGA数据库的分析结果显示,CDKN2A在肝癌中属于不良预后因子并在高危组中高表达。此外通过对TCGA数据库中其它癌症类型的数据分析结果,发现在子宫内膜癌、肾癌中CDKN2A同样属于不良预后因子,类似的结果在Larque A B等[27]关于hpv阴性的喉部鳞状细胞癌的研究中也有所提及。总而言之CDKN2A作为一种重要的抑癌基因,其纯合缺失、启动子甲基化或基因点突变与肿瘤的发生密切相关,但其mRNA表达水平作为肝癌患者预后因子的研究还不充分,CDKN2A是否为肝癌的不良预后因子还需更加系统、深入的研究和验证。FOXO又名叉头蛋白(Forkhead box protein)是一类转录因子,广泛参与到细胞新陈代谢、分化、凋亡、增殖等生命活动中,尤其在细胞周期进程的调控和程序化死亡中起到重要作用。许多研究表明,FOXO的失活与肿瘤的发生发展显著相关,是哺乳动物细胞最重要的一类抑癌基因,并且在多种肿瘤中都可观察到FOXO转录因子的异常表达。FOXO1是叉头转录因子FOXO家族的一个重要成员,FOXO1在肝癌组织中低表达,导致肝癌细胞的细胞周期失控和凋亡异常抵抗,进而加速肿瘤的进展。提高FOXO1的活性,可影响上调细胞周期抑制蛋白p21、p27和下调细胞周期蛋白cyclinD1的表达从而抑制肝癌细胞增殖,并通过激活促凋亡蛋白Bim,诱导肝癌细胞凋亡[28-30]。研究发现,由FOXO1突变引发的mRNA表达水平的变化,对肝癌患者的分子分型起到重要作用同时与预后存在较强的相关性。基于以上结果我们推断,FOXO1可能是肝癌的肿瘤抑制基因,该基因突变导致的表达水平降低可导致肝癌患者生存期缩短。
此外本研究还发现,两种分子分型的差异表达基因功能,与肿瘤细胞侵蚀、转移、复发过程相关的信号通路相关,包括与高危组显著相关的信号通路包括:ECM-受体相互作用,黏附信号通路[31-32],以及与肿瘤细胞增殖相关的PI3K-Akt信号通路[33-34];而低危组中异常通路信号则主要与细胞周期、胰液分泌异常有关。研究表明,肝癌的发病早期往往就出现门静脉侵袭、肝内转移以及肝外肺脏和骨组织的转移,而肝癌的侵袭、转移和术后复发是影响患者预后的主要因素。恶性肿瘤发生发展常常伴有细胞外基质(Extracellular Matrix,ECM)及其细胞表面受体表达的变化[35-36];而黏附信号通路的激活则在细胞分化,发育以及增殖,凋亡方面起重要作用,并参与肿瘤的侵袭,运动和转移过程[37];PI3K/Akt/mTOR信号通路则作为细胞内重要信号传导通路之一,可通过影响下游多种效应分子的活化状态,维持肿瘤细胞恶性增殖的生物学特性[32]。由此可以看出高危组肿瘤的恶性程度高,侵袭性强,极易复发和转移,是造成患者预后较差、生存率较低的主要原因。
利用TCGA数据库中肝癌患者SNP突变数据和转录组芯片数据的联合分析,通过蛋白互作网络筛选出核心基因,并构建肝癌分子分型方法。在没有任何先验信息的情况下对肝癌患者进行分子分型,分析结果表明这种分子分型方法对肝癌患者的预后评估具有一定的作用。另外通过 WGCNA和KEGG富集分析探寻了高危、低危两种分型间的基因表达和异常代谢通路的差异,对造成不同肝癌分子分型患者生存率差异的原因进行进一步研究,同时筛选出的核心基因CDKN2A、FOXO1也可作为肝癌早期诊断,预后监测的新型分子标志物以及分子治疗的新靶点。
4 结 论
1)CDKN2A和FOXO1在肝癌分子分型中起到决定性作用,且CDKN2A高表达和FOXO1低表达可能与肝癌患者的不良预后密切相关。
2)高危组异常信号通路ECM-受体相互作用,黏附信号通路,PI3K-Akt信号通路可能与恶性程度高,侵袭性强,极易复发和转移,预后较差有关。
