高速逆流色谱法从小叶金钱草中分离四种黄酮苷类化合物
2022-12-10宋道光
宋道光,陈 志,
(1.青海师范大学地理科学学院,青海西宁 810008;2.青海师范大学,青藏高原药用动植物资源重点实验室,青海西宁 810008)
小叶金钱草(Hydrocotyle sibthorpioidesLam.)为伞形科天胡荽属植物,中国传统中药之一[1]。可全草入药,具有清热解毒、利湿退黄、跌打淤肿以及抗菌等功效,民间及临床上常用于治疗黄疸型肝炎、结石及胆囊炎等病症,但尚未被国家药食同源性中药材及保健食品目录收录。化学成分方面,文献报道小叶金钱草化学成分主要集中在黄酮及其苷类、萜类、甾醇、氨基酸、挥发油、香豆素类等化合物中[2-3]。药理研究方面,小叶金钱草除具有极低的毒性外[4],具有降血糖[5]、神经保护[6]、抗氧化[7]、抗癌[8]以及肝损伤保护[9]等药理活性。上述药理活性与其中所含的多种生物活性物质密切相关,例如黄酮类、三萜类和挥发油等,其中黄酮类化合物是其中一类非常重要的生物活性物质,在自然界中分布广泛,且具有抗癌、抑菌、抗衰老等多种生物活性[10],能与食品中的一些成分发生相互作用继而影响食品的品质[11],并应用于减肥等保健食品中[12-13]。以上研究表明,金钱草中的黄酮类化合物是一类重要的活性成分。
熊峻等[3]对金钱草及其同属植物中的黄酮类成分进行了归纳总结,但文献中对黄酮类成分的分离研究还局限在柱层析(硅胶、凝胶、MCI)及重结晶等传统手段。为了更好的研究金钱草中的化学成分,通常会借助一些其他手段如响应面法辅助以超声提取,能够优化提取工艺达到更好的提取有效成分的目的[14],此外高效液相色谱(HPLC)[15]、气相色谱质谱(GCMS)[5]、超高效液相色谱质谱(UPLC-MS)[7]也应用于分析金钱草化学成分。因此,借助现代仪器分析手段能更好的对中药中的化学成分进行研究。截至2020版药典,小叶金钱草仍未被收录,因此对小叶金钱草化学成分进行深入研究具有重要意义。
虽然已有诸多学者对金钱草中黄酮类成分进行研究,但大多还局限于传统手段,少有通过高速逆流色谱对金钱草化学成分进行研究的报道,传统植化分离手段大多存在分离时间长、效率低下、溶剂使用量大及部分分离物质死吸附等缺点。高速逆流色谱(High-speed Countercurrent Chromatography,HSCCC)是1982年由美国国立卫生院Ito博士研发的一种新型、连续高效的液液分配色谱技术,即按照某样品在两相不互溶的溶剂之间分配的一种技术[16]。除节约溶剂、缩短分离时间外,还可有效解决传统柱色谱在分离时产生的不可逆吸附以及样品在分离过程中遇到的变性问题[17]。占颖等[18]对高速逆流色谱在黄酮类天然产物的分离应用进行了总结分析,包括适合分离黄酮及其苷类的溶剂系统、分配系数、辅助添加剂、洗脱方式以及与传统分离手段的结合情况等。本研究在前期对小叶金钱草乙酸乙酯萃取物研究的工作基础上,采用HSCCC手段对小叶金钱草正丁醇萃取物中的黄酮苷类成分进行进一步分离研究,通过调整不同溶剂系统,采取梯度洗脱方式并结合前期总结的HSCCC样品出峰经验公式,优化分离条件,为进一步研究小叶金钱草的化学成分提供分离方法及理论依据。
1 材料与方法
1.1 材料与仪器
小叶金钱草 西宁城北百信大药房,青海师范大学陈志教授鉴定为天胡荽属植物天胡荽(Hydrocotyle sibthorpioidesLam.);DM 301型大孔吸附树脂 安徽三星树脂科技有限公司;葡聚糖凝胶 (Sephadex LH-20)、反相硅胶(ODS/RP-18) 安徽博美生物科技有限公司; 正己烷、正丁醇、乙醇、甲酸 分析纯,天津市百世化工有限公司;乙腈 色谱纯,天津星马克科技发展有限公司;水 二次蒸馏水;核磁管(外径5 mm,长7英寸) 美国Wilmad公司;DMSO-d6宁波旋光医药科技有限公司。
EV400旋转蒸发器 北京莱伯泰科仪器股份有限公司; KJ-11030AL型超声波清洗器 深圳市科洁超声科技有限公司; Waters 600高效液相色谱仪、真空脱气过滤装置、Waters 2998 PAD二极管阵列检测器 美国Waters公司; HSCCC TBE-300B型高速逆流色谱仪 中国上海同田生化技术有限公司;N2000双通道色谱工作站 浙江大学智达信息工程有限公司;Bruker-500 MHz NMR(AVANCE III HD)核磁共振仪 瑞士Bruker公司。
1.2 实验方法
1.2.1 小叶金钱草正丁醇萃取物的制备 将购买的小叶金钱草全草粉碎后过40目筛。提取方法参考课题组前期工作[19],称取20 kg药材粉末分次置于KJ-11030AL型超声波清洗器中,放置药材前按液料比10:1加入70%(v/v)乙醇溶液,于600 W、40 kHz、60 ℃条件下超声提取1 h,过滤所得药渣再按以上方式继续提取三次,总计提取4次。过滤液经减压浓缩后得棕褐色黏稠浸膏。浸膏加入蒸馏水20 L并在加热条件下(60 ℃)超声溶解,然后所得溶液依次用10倍体积石油醚、乙酸乙酯、正丁醇萃取5次。浓缩正丁醇萃取液,冻干得正丁醇萃取物,4 ℃避光保存备用。
1.2.2 小叶金钱草正丁醇萃取物各分离样品的制备
取正丁醇萃取物255 g,用5 L蒸馏水超声溶解,DM 301型大孔吸附树脂柱(10×50 cm)进行富集并静置过夜,使用乙醇/水(0:100、30:70、50:50、75:25、100:0)进行洗脱,每个梯度各用五倍柱体积洗脱,20.0 mL/min流速洗脱。收集50%乙醇/水洗脱液,旋蒸除去溶剂并于-70 ℃冷冻干燥24 h,得大孔树脂富集样品。
取大孔树脂富集样品(10 g)加入100 mL蒸馏水溶解并过0.45 μm滤膜,Sephadex LH-20柱(3×100 cm)柱层析分离,分别用甲醇/水(0:100、10:90、20:80、30:70、40:60、50:50、75:25、100:0)洗脱,每个比例洗脱2个柱体积[19],1/20柱体积为一馏分,1.0 mLmin流速洗脱,收集40%甲醇洗脱组分,旋蒸除去溶剂并于-70 ℃冷冻干燥24 h,得Sephadex LH-20分离样品。
Sephadex LH-20分离样品(1 g)加入10 mL蒸馏水超声溶解并过0.45 μm滤膜,ODS柱(2×100 cm)柱层析分离,分别用甲醇/水(0:100、10:90、20:80、30:70、40:60、50:50、75:25、100:0)洗脱,每个比例洗脱2个柱体积,1/20柱体积为一馏分,1.0 mL/min流速洗脱,旋蒸除去溶剂并于-70 ℃冷冻干燥24 h,得ODS分离样品,所得单体化合物经HPLC检测其纯度。
1.2.3 分离样品的高效液相色谱检测 取一定量正丁醇萃取物(20 mg)、大孔树脂富集样品(6.5 mg)、Sephadex LH-20分离样品(4 mg)、ODS分离样品(2 mg),用1 mL乙腈溶解,0.45 μm滤膜过滤后,进行HPLC分析,样品纯度按面积归一化法计算。
色谱柱:Waters 600 SunFireTMC18柱(4.6×250 mm,5 μm),流动相为乙腈-0.1%甲酸(15~30%乙腈,0~70 min),流速为1.0 mL/min,柱温为25 ℃,检测波长254 nm,进样10 μL分析。高效液相色谱(HPLC-PAD)紫外光谱扫描范围:200~400 nm。
1.2.4 高速逆流色谱溶剂系统的选择
1.2.4.1 溶剂系统的确定 参考高速逆流色谱分离黄酮苷类化合物溶剂系统[20-21]。按照溶剂极性的不同,配制乙酸乙酯-正丁醇-水溶剂系统(体积比为1:1:1、2:1:2、4:1:4),正己烷-正丁醇-水溶剂系统(体积比为1:1:1、1:2:2、1:2:1)和正己烷-正丁醇-水-冰乙酸溶剂系统(体积比为1:2:1:0.1、1:1:1:0.1),考察乙酸乙酯、正己烷、正丁醇和冰乙酸对分配系数(k)的影响。
1.2.4.2 k值的测定 k值及分离度(α)的测定方法、参考值范围参照文献[19]。k值定义为:样品溶解在不互溶的两相溶剂中,上相中目标化合物的色谱峰面积(S上)除以下相中目标化合物的色谱峰面积(S下)(k=S上/S下),其中k的范围为0.2~5.0,α的定义为α=k1/k2(k1>k2),且α>1.5。
1.2.5 高速逆流色谱法分离制备小叶金钱草中的黄酮苷 配制溶剂系统I:正己烷-正丁醇-水-冰乙酸(1:2:1:0.1,v/v/v/v)和系统II:正己烷-正丁醇-水-冰乙酸(1:1:1:0.1,v/v/v/v),于分液漏斗中充分振摇后在室温下静置过夜,分别将系统I、系统II的上下相分离并超声脱气20 min,以系统I的上相为固定相,系统I的下相为流动相。仪器参数如下:循环水温度45 ℃,主机正转模式,转速900 r/min,流动相流速2 mL/min,检测波长为254 nm,其余操作步骤同文献[19]。固定相保留率(SF)计算方法为:SF(%)=[(螺旋管体积-流出固定相体积)/螺旋管体积]×100。其中溶剂系统I固定相保留率为,SF1(%)=[(300-110)]/300×100=63.3%,系统II的固定相保留率SF2(%)=[(300-115)]/300×100=61.6%。
开启TBD 2000检测系统,设置采集数据。取1.2.2中ODS分离样品324 mg,用10 mL系统I上相液体溶解,由进样阀进样,记录HSCCC色谱图并收集分离组分。当第一组色谱峰流出检测器后,上相固定相不变,下相流动相迅速切换为系统II的下相作为流动相,继续洗脱,继续收集分离组分。
1.2.6 化合物核磁共振波谱表征 取单体化合物各10 mg,用0.5 mL DMSO-d6溶解,移入核磁管。通过核磁共振波谱仪对已分离化合物进行波谱鉴定(1H NMR、13C NMR)。1H NMR参数:扫场范围=-3.8247~16.1698 ppm,TD=65536,NS=8,DS=2,SWH=10000.000 Hz,FIDRES=0.152588 Hz,AQ=3.2767999 sec,RG=106.4,DW=50.000 usec,DE=6.50 usec,TE=298.0 K,D1=1.00000000 sec;13C NMR参 数:扫 场 范 围=-18.3314~218.3255 ppm,TD=65536,NS=160,DS=4,SWH=29761.904 Hz,FIDRES=0.454131 Hz,AQ=1.1010048 sec,RG=192.1,DW=16.800 usec,DE=6.50 usec,TE=298.0 K,D1=2.00000000 sec,D11=0.03000000 sec。四甲基硅烷(TMS)定标。
1.3 数据处理
1H NMR、13C NMR图谱在MestReNova 6.1.0软件中处理,并进行1H NMR化学位移积分和计算偶和常数(J),J的计算方法参照文献[19]。通过与文献中化合物核磁数据进行比对,比对一致即确定化合物结构,结构式通过ChemBioDraw 14.0画出。
2 结果与分析
2.1 小叶金钱草各分离样品HPLC分析
按照1.2.3方法分析样品,正丁醇萃取物、DM 301大孔吸附树脂富集样品、Sephadex LH-20分离样品以及ODS分离样品的HPLC结果分别为图1a~图1d。由图1a可知,正丁醇萃取物中大极性杂质较多导致其他组分色谱峰并不明显。而由图1b可知,正丁醇萃取物经DM 301大孔树脂富集后,主要成分被明显富集,但所包含的其它杂质仍较多且复杂。经Sephadex LH-20进行纯化后分析结果见图1c,可知经前期萃取富集和柱层析纯化后,4个主要成分进一步得到纯化,其保留时间分别为:22.859,29.109,34.157和42.910 min。由1.2.3所述的色谱条件可知,正丁醇萃取物主要成分极性较大,因此大孔吸附树脂富集样品样品经Sephadex LH-20纯化后,选择ODS进行进一步分离,得到两种单体化合物2、3和一组混合物,即ODS分离样品。单体化合物纯度分析见2.4,ODS分离样品分析见图1d,峰1、峰4为高速逆流色谱目标分离成分。
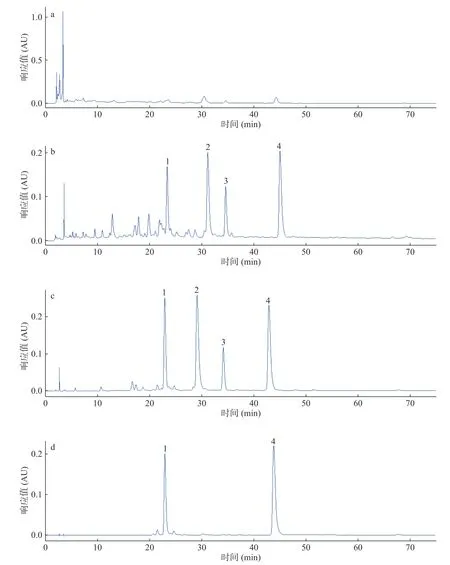
图1 不同萃取物的HPLC色谱图Fig.1 HPLC chromatograms of different fractions
2.2 HSCCC溶剂系统的优化及确定
2.2.1 溶剂系统的确定 不同逆流色谱溶剂系统的k值变化如表1。根据HPLC分析及试验发现,ODS分离样品中的待分离目标化合物极性较大且易溶于正丁醇和水,而逆流色谱中包含正丁醇的溶剂系统常被用于分离大极性化合物[22]。从表1可以看出,在包含乙酸乙酯的溶剂系统中(系统1、2、3),4号化合物的k均较大,不符合k的最适范围, 另一方面,包含乙酸乙酯的溶剂系统具有更长的两相分层时间,在HSCCC分离过程中两相溶剂分层时间过长将不利于分离效果,因此不适合用作HSCCC分离的溶剂系统。

表1 ODS分离样品在不同溶剂体系中的分配系数Table 1 Partition coefficients(K) of the reversed-phase silica gel sample in different two phase solvent systems
当用正己烷替换溶剂系统中乙酸乙酯后(系统1和4),化合物的分配系数明显减小而分离度明显增加。当正己烷比例减小或正丁醇比例增加时(系统4与5、6),化合物分配系数增加而分离度减小,但1号化合物的k太小而不利于分离,因此系统4、5、6不适合用作HSCCC分离的溶剂系统。据文献报道,Chen等[23]向溶剂系统中添加冰乙酸来改善化合物的分离。 因此在正己烷-正丁醇-水溶剂系统中加入冰乙酸来改善1号和4号化合物的分离,进一步探索正己烷-正丁醇-水-冰乙酸溶剂比例。
2.2.2 正己烷-正丁醇-水-冰乙酸比例的确定 表1中(系统4和7,系统6和8)可以发现,溶剂系统中添加冰乙酸对化合物的分离度几乎没有影响,但却有效地增加了目标化合物的分配系数。研究发现,系统7(系统I)最适合分离1号化合物,系统8(系统II)最适合分离4号化合物。单一的一种溶剂系统难以做到同时1号和4号化合物,而HSCCC梯度洗脱方式能在确保1号化合物纯度的情况下,在最短时间内分离并节省4号化合物的分离时间,避免目标化合物过早的与杂质流出以确保化合物的纯度,因此选取上述两种溶剂系统并以梯度洗脱方式分离目标化合物。
2.3 HSCCC分离结果
参照1.2.5中并对ODS样品进行高速逆流色谱分离,得到2个分离组分(图2)并进行HPLC分析。收集图2中88~102 min(组分I)及310~330 min(组分II)两组分进行HPLC分析,HPLC分析结果见2.4。
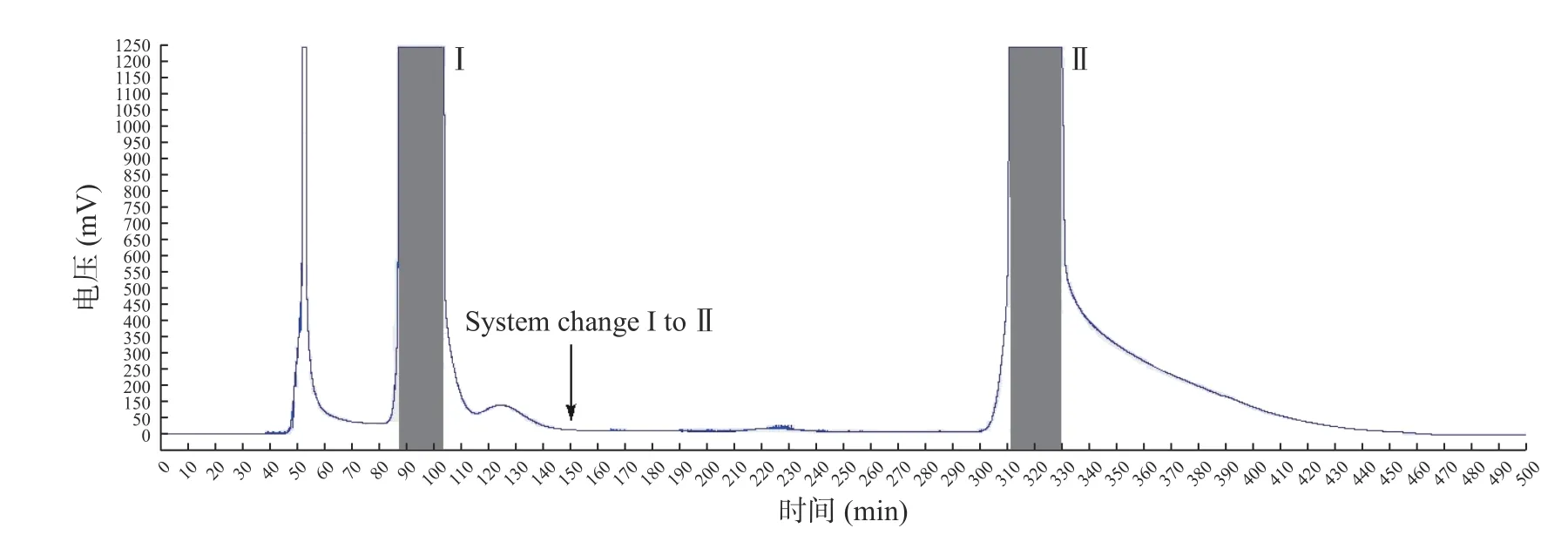
图2 ODS分离样品的高速逆流色谱图Fig.2 HSCCC c hromatogra m of th e OD S s ample
2.4 单体化合物HPLC分析
按1.2.3的HPLC方法分析样品,HSCCC中组分I对应图3a中1号化合物,Rt=22.859 min,纯度97.7%,对应图1中1号色谱峰。HSCCC中组分II对应图3d中4号化合物,Rt=42.910 min,纯度95.3%,对应图1中4号色谱峰。2.1中经ODS分离得到的两个单体化合物分别对应图3b中2号化合物,Rt=29.109 min,纯度96.8%,即图1中2号色谱峰。图3c中3号化合物,Rt=34.157 min,纯度95.7%,对应图1中3号色谱峰。将柱层析与高速逆流色谱所分离得到化合物经冷冻干燥后称重得1号化合物(100.23 mg)、2号化合物(150.29 mg)、3号化合物(128.43 mg)和4号化合物(105.49 mg)。各色谱图中小图对应各色谱峰的紫外吸收光谱,由紫外光谱分析得知,色谱峰紫外吸收在峰带I,300~400 nm及峰带II,220~280 nm有明显紫外吸收,符合黄酮类化合物紫外光谱吸收特征,故所分离得到的化合物为黄酮类化合物[24]。
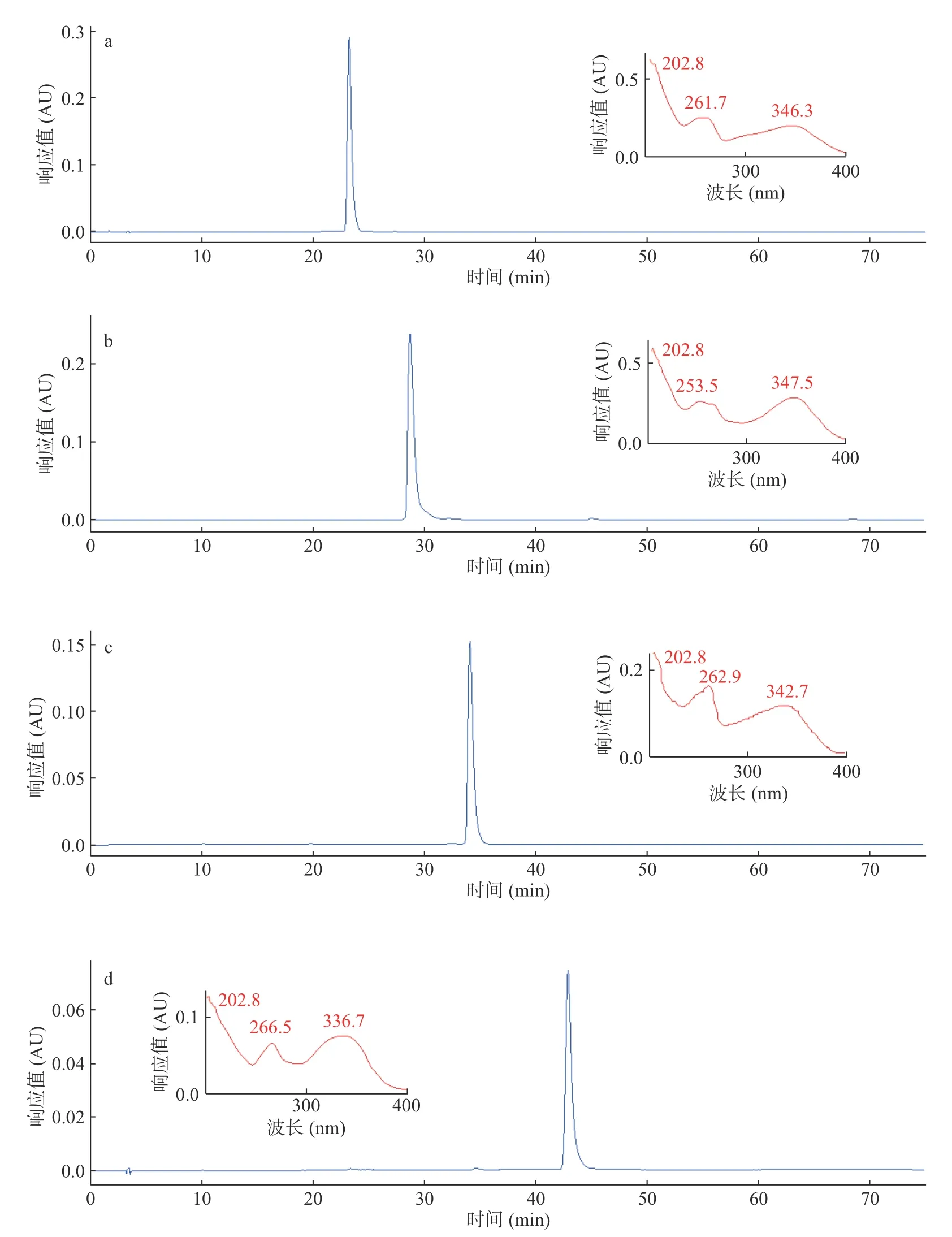
图3 HSCCC及反相硅胶分离化合物的HPLC色谱图Fig.3 HPLC chromatograms of the targeted compounds purified by HSCCC and reversed-phase silica gel.
2.5 高速逆流色谱出峰经验公式
为了验证HSCCC梯度洗脱是否真正起到了缩短分离时间的作用,对课题组前期实验进行归纳,总结高速逆流色谱出峰经验公式,当k≥1时,有如下公式:
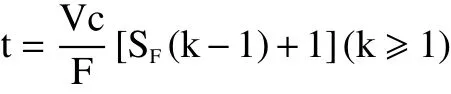
式中,t为高速逆流色谱色谱峰出峰时间(min);Vc为螺旋管体积(mL);F为流动相流速(mL/min);SF为固定相保留率(%);k为分配系数。
据此公式1估算溶剂系统I中化合物4的出峰时间,t=300/2[0.633(4.68-1)+1]=499.416 min,小于化合物4实际分离时间(约300~310 min)。根据系统I切换系统II的时间(150 min),以此为基点计算系统II条件下,估算化合物4的出峰时间。t=300/2[0.616(1.17-1)+1]=165.708 min,以及基点时间150 min预计分离时间315 min左右,与实际分离时间较为接近。
2.6 结构表征
1、2和4号化合物的1H NMR和13C NMR信息与前期实验工作所得化合物信息相同[25],鉴定为杨梅素3,3'-二-α-L-鼠李糖苷(1号化合物)、木犀草素-7-O-β-D-葡萄糖醛酸苷(2号化合物)和芹菜素-7-O-β-D-葡萄糖醛酸苷(4号化合物)。化合物3前期实验尚未分离得到,核磁信息如下,1~4号化合物结构式见图4。
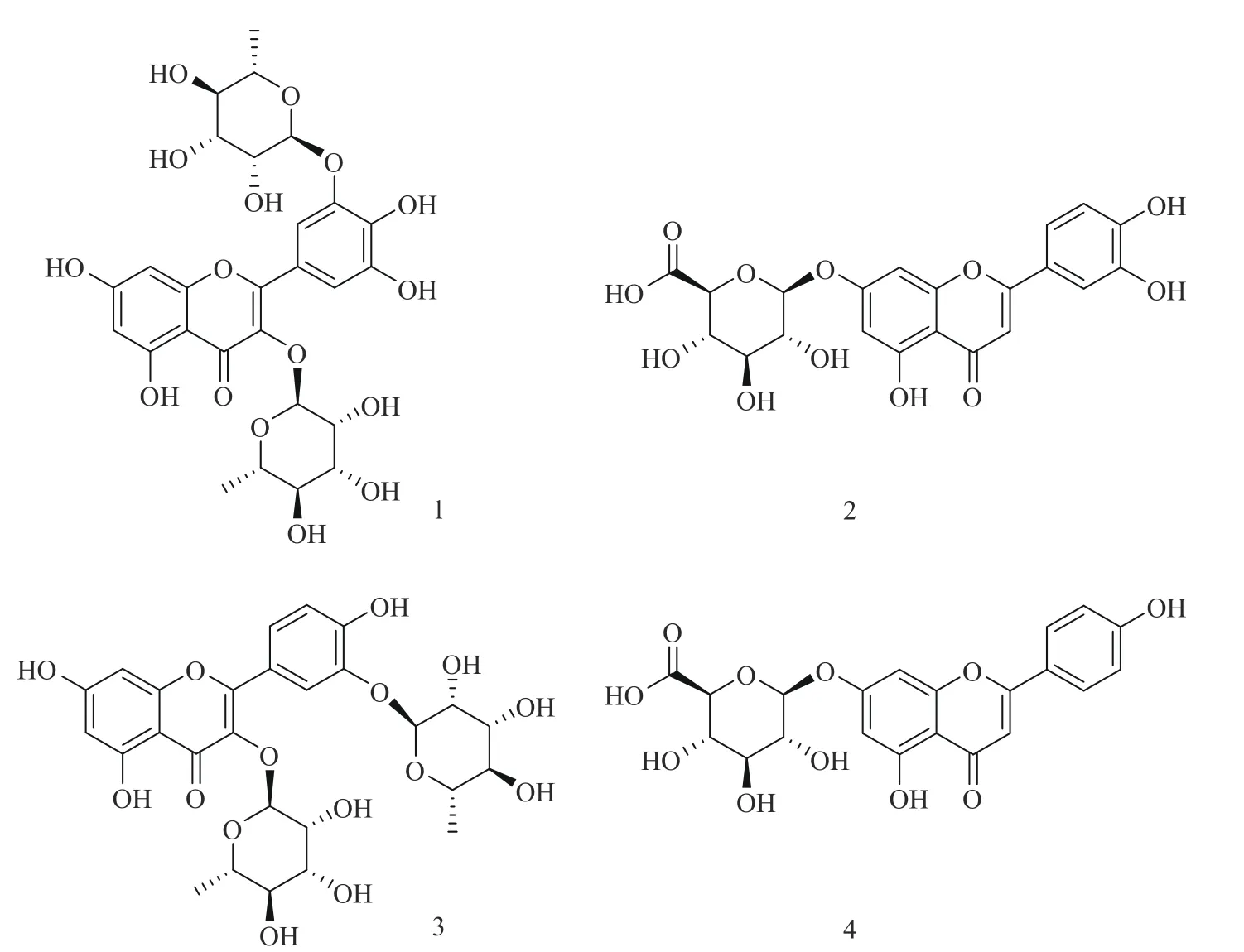
图4 1~4号化合物的化学结构Fig.4 Chemical structure of Compound 1~4
3号化合物:1H NMR(DMSO-d6,500 MHz,ppm):δ7.53(1H,d,J=2.1 Hz,2'-H),7.46(1H,dd,J=2.1 Hz,2.1 Hz,6'-H),6.97(1H,d,J=8.4 Hz,5'-H),6.27(1H,d,J=1.9 Hz,8-H),6.11(1H,d,J=1.9 Hz,6-H),5.30(1H,s,3-rha-1-H),5.26(1H,s,3'-rha-1-H),3.47-3.97(4H,m,3-rha-2,3,4,5),3.11-3.96(4H,m,3'-rha-2,3,4,5),1.17(3H,d,J=6.2 Hz,3'-rha-CH3),0.81(3H,d,J=6.2 Hz,3-rha-CH3)。13C-NMR(DMSO-d6,125 MHz,ppm):δ177.70(C-4),167.84(C-7),161.65(C-5),157.16(C-8a),156.81(C-2),144.78(C-3'),116.76(C-5'),119.35(C-2'),151.79(C-4′),134.38(C-3),121.05(C-1'),125.25(C-6'),103.45(C-4a),102.05(C-1''),100.95 (C-1'''),99.97(C-6),94.46(C-8),72.41(C-4''),71.63(C-4'''),71.04(C-3''),70.86(C-3'''),70.80(C-2''),70.60(C-2'''),70.49(C-5''),70.06(C-5'''),18.38(C-6'''),17.95(C-6'')。通过与文献[26]对比,鉴定化合物3为槲皮素3,3'-二-α-L-鼠李糖苷(Quercetin 3,3'-di-α-L-rhamnopyranoside)。
从化合物的来源来看,上述四种化合物在其他植物中也有报道,其中化合物1、3只在大金钱草(Lysimachia christinaeHance)中[26]分离得到,目前还没有关于这两种化合物活性方面的报道。化合物2和4除在前期工作中分离得到外,也广泛分布在其他植物中,如穿心莲(Andrographis paniculata(Burm.f.)Nees.)[27]、鹅不食草(Centipeda minima(L.)A.Br.et Aschers.)[28]及独脚金(Striga asiatica(L.)O. Kuntze)[29]等。活性方面化合物2和4具有抗氧化活性、降糖活性[30]、抗心肌缺血活性[31]、抗癌及抗炎[32]等药理活性。而从小叶金钱草中分离得到的黄酮苷类成分则进一步丰富了上述四种化合物的植物来源,为进一步研究药理活性奠定了基础。
3 结论
本文采用柱层析与高速逆流色谱结合的方式分离制备小叶金钱草中四种黄酮苷类成分,以正己烷-正丁醇-水-冰乙酸(系统I:1:2:1:0.1,v/v/v/v)及正己烷-正丁醇-水-冰乙酸(系统II:1:1:1:0.1,v/v/v/v)为溶剂系统,在转速900 r/min、流速2 mL/min的条件下,以梯度洗脱方式分离制备分配系数差异较大的黄酮苷类化合物,并辅以高速逆流色谱出峰经验公式验证洗脱效果。分离化合物经1H NMR和13C NMR鉴定为杨梅素3,3'-二-α-L-鼠李糖苷(1)、木犀草素-7-O-β-D-葡萄糖醛酸苷(2)、槲皮素3,3'-二-α-L-鼠李糖苷(3)和芹菜素-7-O-β-D-葡萄糖醛酸苷(4),纯度均高于95%,其中化合物3为首次从小叶金钱草中分离得到,为进一步研究小叶金钱草的化学成分、药效以及综合利用奠定了理论基础。
