解吸附电晕束电离质谱法快速测定黄精中的薯蓣皂苷元
2022-12-09陈诗丽谢小玉
陈诗丽,闵 可,杨 蓉,罗 玮,谢小玉,陈 波,马 铭
(湖南师范大学化学生物学及中药分析教育部重点实验室,植化单体开发与利用湖南省重点实验室,中国 长沙 410081)
黄精中薯蓣皂苷元含量的测定方法主要有紫外分光光度法(UV)[6]、高效液相色谱-紫外检测法(HPLC-UV)[7]和超高效液相色谱-紫外检测法(UPLC-UV)[8]。紫外分光光度法虽然操作简单,但分析复杂体系时,分析物易受共存物的干扰。由于薯蓣皂苷元没有发色团,紫外吸收较弱,且紫外吸收波长主要在低波长区域,因此通过UPLC/HPLC-UV法检测时灵敏度较低。此外,当采用HPLC-UV或UPLC-UV法对薯蓣皂苷元进行定量时,均需要繁琐的样品预处理过程和耗时的色谱分离过程,不适用于高通量分析。因此,迫切需要建立一种高效、灵敏、快速和简单易操作的黄精中薯蓣皂苷元的检测方法。
敞开式质谱是一种可以在常压下直接分析样品或样品表面的技术,具有快速、灵敏、实时、高通量,以及适用于复杂样品分析的特点,已被用于食品检测[9]和临床检验[10,11]。解吸附电晕束电离质谱法(DCBI-MS)是一种新型的敞开式质谱分析技术,样品无需前处理,分析过程仅需十几秒,可直接分析固体、液体和粉末样品[12]等。与其他敞开式质谱技术相比,DCBI源可以产生可见的紫色电晕束,有利于样品的原位分析和质谱成像[13]。Min等人[14]使用DCBI-MS/MS法快速原位高通量分析了口罩中的邻苯二甲酸酯。此外,DCBI-MS法已被广泛应用于食品安全、包装材料和有机农药分析等领域[15,16]。
本研究建立了DCBI-MS法测定中药黄精中的薯蓣皂苷元的方法,根据质谱准分子离子峰和碎片离子对薯蓣皂苷元进行快速识别,并对其裂解规律进行探究。
1 仪器与材料
1.1 仪器
FW-80高速万能粉碎机(北京市永光明医疗仪器有限公司)、AP135W电子天平(日本岛津公司)、MTN-2800D氮吹浓缩装置(天津奥特赛恩斯仪器有限公司)、J03-0.2A精密手动压力机(上海申康机床有限公司)、F-030SD超声波清洗机(深圳福洋科技集团有限公司)、DCBI-1电离源(日本岛津公司)、LCMS-8040质谱仪(日本岛津公司)和HPLC-8050高效液相色谱仪(日本岛津公司)。
1.2 材料
甲醇、乙腈和二氯甲烷(均为色谱级)购自北京伊诺凯科技有限公司(中国,北京)。实验用水使用超纯水系统(Millipore,美国)制得。薯蓣皂苷元标准品(批号:Z10S11B124158)购自上海源叶生物科技有限公司(中国,上海)。甲睾酮标准品(内标)购自萨恩化学科技有限公司(中国,上海)。黄精药材购买途径来自湖南长沙市各大药房及电商中药材店铺,信息如表1所示。黄精药材经湖南师范大学医学院邹辉副教授鉴定为黄精Polygonatirhizoma的干燥根茎。

表1 黄精样品信息
2 方法
2.1 DCBI-MS工作原理与条件

图1 DCBI-MS原理示意图
使用DCBI-1电离源与LCMS-8040质谱仪结合,在选择离子监测(SIM)的正离子模式下工作。DCBI-MS的原理图如图1所示。高压氦气经过控制加热套管加热后,经空心针电极(工作电极)射出,在工作电极和环形电极(也称为对电极)形成的强电场作用下,被激发电离形成等离子体[17]。等离子体伴随气流流出DCBI源探头产生可见的紫色电晕束。将样品溶液滴加在制备好的三角形纸基上,样品被电晕束照射后,分析物被解吸附,经过一系列电离反应,形成带电离子进入质谱锥孔被检测。氦气(He)流量为1.0 L·min-1,电压设定为1.8 kV,解吸附温度为320 ℃。MS界面由LabSolutions工作站控制,条件为:脱溶剂(DL)温度为250 ℃;加热块温度为350 ℃;雾化气流量为0.5 L·min-1。
2.2 HPLC-UV条件
色谱分离采用Diamonsil C18(2)色谱柱(250 mm×4.6 mm,5 μm)。流动相为水(A)-乙腈(B),等度洗脱条件为V(乙腈)∶V(水)=94∶6,总流速设置为1.0 mL·min-1,检测波长为210 nm,进样体积为20 μL,单次色谱分析总时间为30 min。
2.3 溶液的制备
2.3.1 对照品溶液的制备 准确称取2.0 mg薯蓣皂苷元标准品于烧杯中,加入甲醇溶解,转至50 mL容量瓶中,定容,得到40 mg·L-1薯蓣皂苷元对照品储备液。内标甲睾酮对照品储备液(40 mg·L-1)的配制方法同上。薯蓣皂苷元对照品储备液用甲醇稀释至10 mg·L-1作为工作溶液,用于优化DCBI-MS的分析参数。
漫长的暴风骤雨之后,被男人浸润透了的伍亦苒满足而又嗔怪地说,怕了你了,像一个饥饿的孩子,房间都订好了,却还要在这里。
2.3.2 供试品溶液的制备 中药黄精药材用高速万能粉碎机粉碎成粉末,过0.45 mm筛。称取0.4 g黄精粉末于离心管中,加入2.00 mL二氯甲烷,静置20 min,在常温下超声40 min,得黄精提取液。对黄精提取液进行两种方式处理:(1)取100 μL黄精提取液,加入甲睾酮内标,用甲醇稀释至溶液总体积为300 μL,得到含内标浓度为2 mg·L-1的供试品溶液,用于DCBI-MS分析。(2)另取1.00 mL黄精提取液,氮吹至干,加入等体积的甲醇复溶,经0.22 μm尼龙膜过滤,得到用于HPLC分析的供试品溶液。
3 结果与讨论
3.1 三角形纸基上样方向的考察
DCBI-MS上样使用的三角形纸基是通过定制的精密手动压力机从色谱纸上裁剪下来的,三角纸的顶角角度为30°,底边宽7 mm,高为13 mm。将待测样品溶液滴在三角形纸基上,笔者考察了三角形纸基底边对着质谱锥孔和三角形纸基尖端对着质谱锥孔两种上样方式对分析物薯蓣皂苷元质谱峰面积响应的影响,结果如图2。由图2可知,当三角形纸基尖端对着质谱锥孔时(图2a),在全扫谱图中背景较复杂,监测不到薯蓣皂苷元的准分子离子[MDiosgenin+H]+(m/z415.3)峰。而当三角形纸基底边对着质谱锥孔时(图2b),背景较为干净,质谱图中出现明显的薯蓣皂苷元的准分子离子峰[MDiosgenin+H]+(m/z415.3),薯蓣皂苷元的质谱响应强度得到了明显提高。推测可能的原因是当三角形纸基底边对着质谱锥孔时,电晕束作用于纸基的表面积更大,有利于更多样品分子进行解吸附和电离。因此,后续采用三角形纸基底边对着质谱锥孔的上样方式开展研究。

图2 上样方式对薯蓣皂苷元质谱响应强度的影响(a)三角纸尖端对着质谱锥孔;(b)三角纸底边对着质谱锥孔
3.2 DCBI-MS条件优化
3.2.1 扫描模式的选择 利用质谱进行定量时,可选择的扫描模式有选择离子监测模式(SIM)和多反应监测模式(MRM)。SIM模式指的是监测薯蓣皂苷元质谱产生的准分子离子[MDiosgenin+H]+的响应强度。MRM模式指的是监测薯蓣皂苷元的一对离子对的响应强度(母离子m/z415.3,子离子m/z217.2)。比较两种模式下目标峰的响应强度,结果如图3。由图3可知,SIM模式下的薯蓣皂苷元的响应强度明显大于MRM模式。此外,SIM模式比MRM模式更简单,不需要离子碰撞诱导解离过程。因此,后续研究均选择SIM模式进行检测。
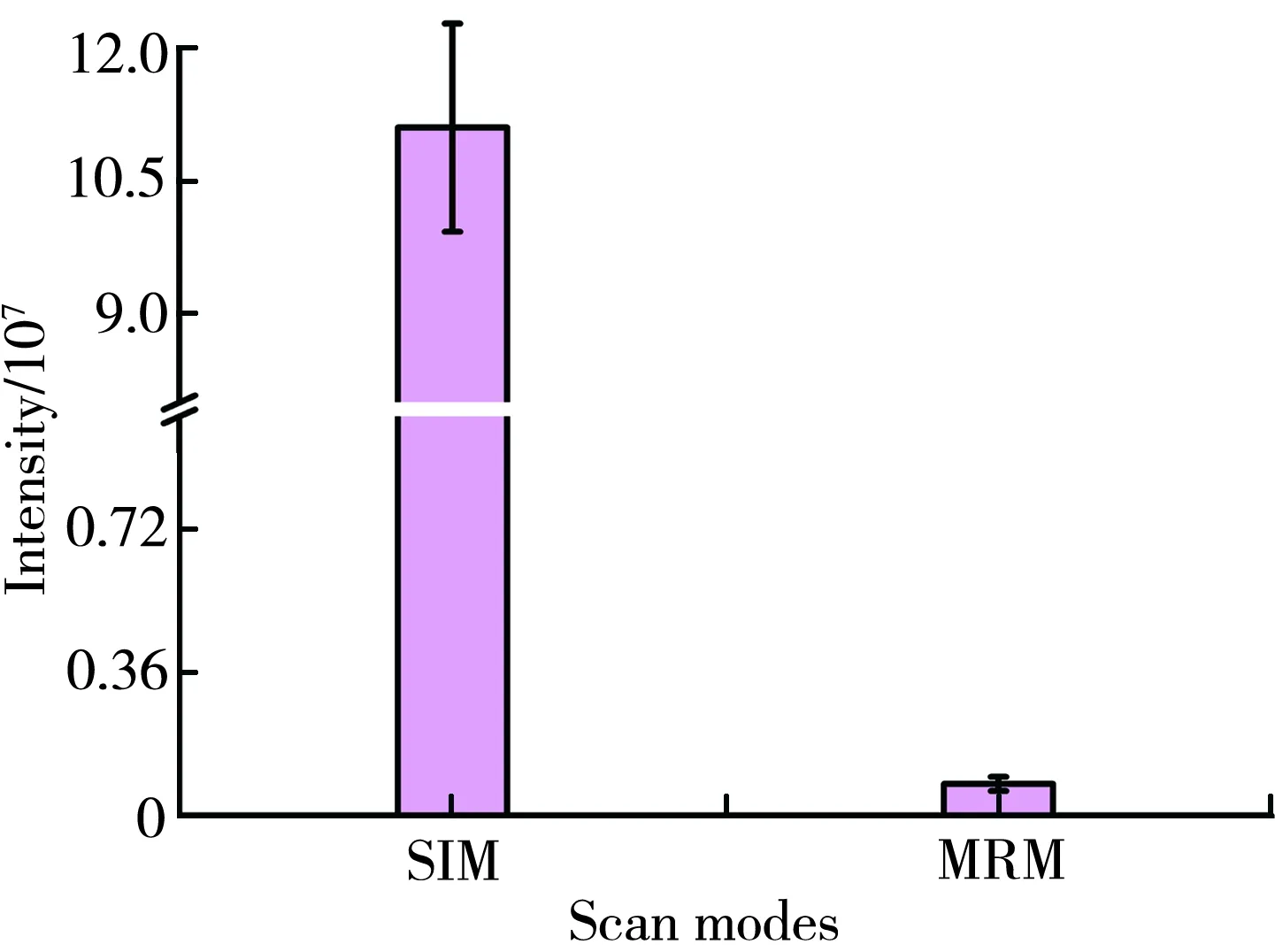
图3 扫描模式对薯蓣皂苷元质谱响应强度的影响
3.2.2 上样体积的影响 在DCBI-MS分析中,样品的上样体积与定量准确性直接相关。一般而言,上样体积与热解吸附出的目标分子总量呈正相关,而与样品的解吸附速度呈负相关,这是由于随着上样体积的增大,上样分析物的物质的量增加,有利于增大分析物的质谱峰面积;但上样体积过大,会降低热传导效率,反而会影响分析物的解吸附及离子化,从而降低分析物的质谱峰面积[18]。笔者考察了薯蓣皂苷元上样体积依次为3,5,10,15,20和25 μL时,薯蓣皂苷元质谱峰面积的变化情况,结果如图4a。图4a表明,上样体积从3 μL增大至20 μL时,分析物的质谱峰面积呈现逐渐增大的趋势,但当上样体积超过20 μL时,分析物的质谱峰面积反而下降,此时,可观察到样品溶液在三角纸上出现轻微溢出现象,说明样品过载造成了部分样品损失。同时,由于上样体积过大,降低了热传导效率,这两个因素共同作用,使得分析物的质谱峰面积出现了显著降低。因此,在后续的研究中,选择上样体积为20 μL。
3.2.3 解吸附温度的影响 DCBI源的电离过程可以分为两步:第一步是样品由凝聚态解吸附成为气相状态;第二步是离子化过程[17]。解吸附温度的升高可以提高解吸附效率,但解吸附温度过高也可能导致分析物分解而造成损失。因此,解吸附温度的选择在DCBI-MS检测中起着至关重要的作用。笔者考察了解吸附温度分别为120,160,200,240,280,320和350 ℃时对薯蓣皂苷元质谱峰面积的影响,结果如图4b。图4b显示,解吸附温度较低时,无法观察到薯蓣皂苷元的质谱信号。解吸附温度从160 ℃逐渐增加到320 ℃时,薯蓣皂苷元的质谱峰面积随着解吸附温度的升高而增加。当解吸附温度超过320 ℃时,薯蓣皂苷元的质谱峰面积反而呈现出了下降的趋势,这可能是由于解吸附温度过高时,薯蓣皂苷元部分分解所致。因此,DCBI源的解吸附温度选择为320 ℃。
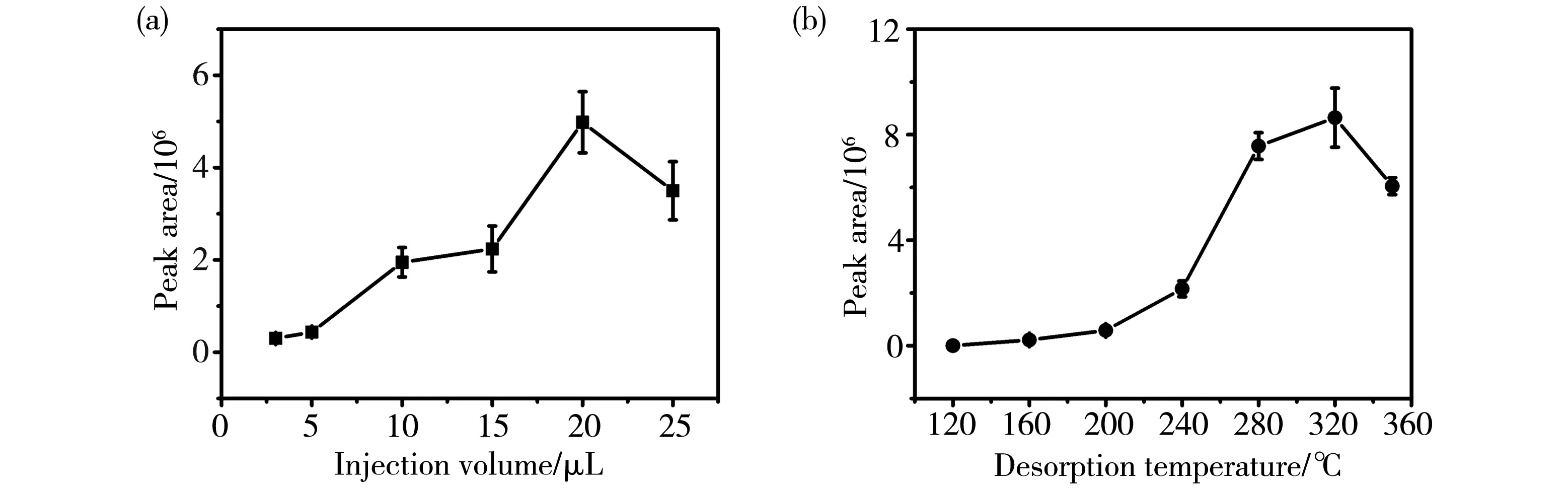
图4 上样体积(a)和解吸附温度(b)对薯蓣皂苷元质谱峰面积的影响
3.2.4 放电电压的影响 相较于其他敞开式质谱,DCBI源的独特优势就在于其拥有一束可见的紫色电晕束,这有利于对样品进行原位分析,进而提高进样位置的定位准确率和质谱成像。为了在保持足够强度的同时获得相对稳定明亮的电晕束,对放电电压进行优化是非常必要的。本文考察了放电电压分别为1.5,1.6,1.7,1.8和1.9 kV时,对薯蓣皂苷元质谱峰面积的影响,结果如图5所示。图5说明,当放电电压从1.5 kV增加到1.8 kV时,分析物的质谱峰面积逐渐增加,同时肉眼观察到电晕束的颜色逐渐加深。当放电电压高于1.8 kV时,薯蓣皂苷元质谱峰面积降低。因此,后续实验选取1.8 kV的放电电压。
3.3 DCBI-MS方法学考察
3.3.1 线性相关性考察 取薯蓣皂苷元对照品储备液,甲醇稀释至质量浓度依次为0.5,3,6,8和10 mg·L-1的一系列标准溶液,不同浓度的标准溶液均含有2 mg·L-1甲睾酮内标溶液。以薯蓣皂苷元浓度为横坐标(X),薯蓣皂苷元与甲睾酮的质谱峰面积的比值为纵坐标(Y),绘制标准曲线(如图6所示),进行线性回归,结果显示,薯蓣皂苷元在0.5~10 mg·L-1时,薯蓣皂苷元与甲睾酮的质谱峰面积的比值与薯蓣皂苷元的质量浓度呈现良好的线性关系,线性回归方程为:Y=0.285 9X+0.047 0,线性相关系数R2为0.999 3。

图5 放电电压对薯蓣皂苷元质谱峰面积的影响
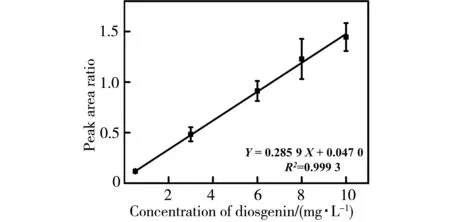
图6 DCBI-MS法测定薯蓣皂苷元校准曲线
3.3.2 精密度、检出限和定量限考察 取10 mg·L-1薯蓣皂苷元工作溶液(含有2 mg·L-1甲睾酮内标),采用DCBI-MS法连续测定3次,记录薯蓣皂苷元准分子离子峰的峰面积与内标峰面积的比值,计算质谱峰面积比值的相对标准偏差(RSD),得到RSD值均为15%,表明仪器精密度良好。检出限(LOD)和定量限(LOQ)分别在信噪比S/N=3和10时测定,连续测定5次,薯蓣皂苷元的LOQ值为0.4 mg·L-1,LOD值为0.1 mg·L-1,表明DCBI-MS法对薯蓣皂苷元有较好的灵敏度。
3.3.3 加标回收率试验 为了验证DCBI-MS法定量检测黄精中薯蓣皂苷元的准确性,须进行加标回收试验。在3份黄精粉末样品(各0.4 g)中分别加入低、中、高浓度水平(即30.0, 75.0和120.0 μg·g-1)的标准溶液,按照“供试品溶液的制备”方法制备加标样品供试品溶液,进行DCBI-MS分析,平行分析5次,测得的加标回收率结果列于表2。由表2可知,加标回收率范围为90.0%~106.8%,表明建立的方法具有良好的准确度。

表2 DCBI-MS法的加标回收率
3.4 方法对比

图7 薯蓣皂苷元对照品溶液与黄精提取物的HPLC图谱
3.4.1 HPLC-UV方法的建立 采用HPLC-UV法对制备得到的黄精提取液进行分析,薯蓣皂苷元工作溶液(10 mg·L-1)与实际样品黄精提取物的HPLC图谱如图7。图7显示,薯蓣皂苷元对照品与实际样品黄精提取物在相同保留时间25.6 min出现了薯蓣皂苷元的特征色谱峰。
用甲醇将薯蓣皂苷元对照品储备液分别稀释至1, 5, 10, 20和30 mg·L-1的系列标准溶液,以薯蓣皂苷元质量浓度为横坐标(X),薯蓣皂苷元的色谱峰面积为纵坐标(Y),得HPLC-UV法线性回归方程,结果表明,薯蓣皂苷元在1~40 mg·L-1时,薯蓣皂苷元色谱峰面积与质量浓度呈现良好的线性关系,线性回归方程为:Y=5 440.8X-1 161.61,相关系数R2为0.999 9。
3.4.2 DCBI-MS法和HPLC-UV法测定结果对比 在本研究中,为了评估DCBI-MS法定量测定黄精中薯蓣皂苷元的准确性,将DCBI-MS法测定的结果与HPLC-UV法测定的结果进行了对比,结果见表3。
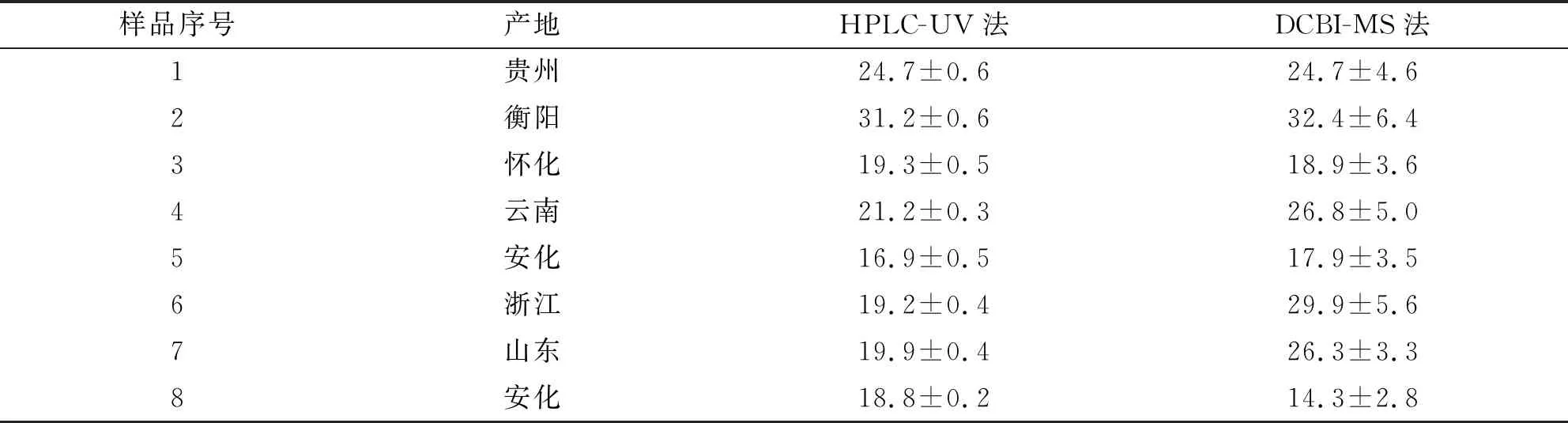
表3 DCBI-MS和HPLC-UV法测定8批黄精样品中薯蓣皂苷元的结果比对 单位:μg·g-1
表3显示,测定的8批次不同产地的黄精药材样品中薯蓣皂苷元的含量为14.3~32.4 μg·g-1。对两种方法的结果进行t检验,显示两组数据之间不存在显著性差异(P>0.05)。值得注意的是,DCBI-MS法进行样品测定时,由于样品基质复杂,样品测定结果的RSD略高于标准溶液测定结果的RSD,但样品测定结果的RSD均在20%以内,符合敞开式质谱的要求,说明DCBI-MS法的测定结果准确可信。从表3可以发现,不同产地黄精的薯蓣皂苷元含量有差异,这可能与黄精种植的土壤、气候和环境等有关。与DCBI-MS法相比,HPLC-UV法需要消耗大量有机流动相,单个样品的分析时长需要30 min。此外,繁琐的前处理过程以及流动相制备过程也需要耗费大量时间。而DCBI-MS法不需要复杂的预处理,样品制备完毕即可上样分析,大大减少了有机流动相的使用,并且20 s内即可分析一个样品,节约了样品分析的时间,具有快速、高效、省时和省溶剂的优势。
3.5 薯蓣皂苷元的裂解规律
将DCBI源与三重四极杆质谱串联,利用二级质谱获得的特征离子对薯蓣皂苷元的裂解规律进行探究。首先,在优化后的最佳分析条件下,正离子模式下得到薯蓣皂苷元的准分子离子峰[MDiosgenin+ H]+(m/z415.3),然后通过碰撞诱导解离(CID)将[MDiosgenin+ H]+离子打碎,得到典型二级质谱图如图8。

图8 薯蓣皂苷元的典型DCBI-MS/MS谱图
从图8可以看出,薯蓣皂苷元的母离子[MDiosgenin+H]+经打碎后主要产生m/z397.3,m/z271.2和m/z253.2的碎片离子峰。借鉴文献报道的薯蓣皂苷的裂解规律[19],根据得到的特征碎片离子,推测薯蓣皂苷元的潜在质谱裂解规律如图9所示。图9显示,薯蓣皂苷元的质子形成[MDiosgenin+ H]+(m/z415.3)的准分子离子峰,m/z415.3离子在碰撞电压的轰击下有两条裂解途径。第一条裂解途径是失去1分子H2O产生 [MDiosgenin-H2O+H]+碎片离子(m/z397.3),接着丢失EF环脱144 Da生成[MDiosgenin-EF-H2O+H]+离子(m/z253.2)。第二条裂解途径是先丢失EF环生成[MDiosgenin-EF +H]+离子(m/z271.2)后再继续丢失H2O脱18 Da产生[MDiosgenin-EF-H2O+H]+离子(m/z253.2)。薯蓣皂苷元潜在裂解规律的发现,对含薯蓣皂苷元的中药材中化合物的鉴别具有指导意义。
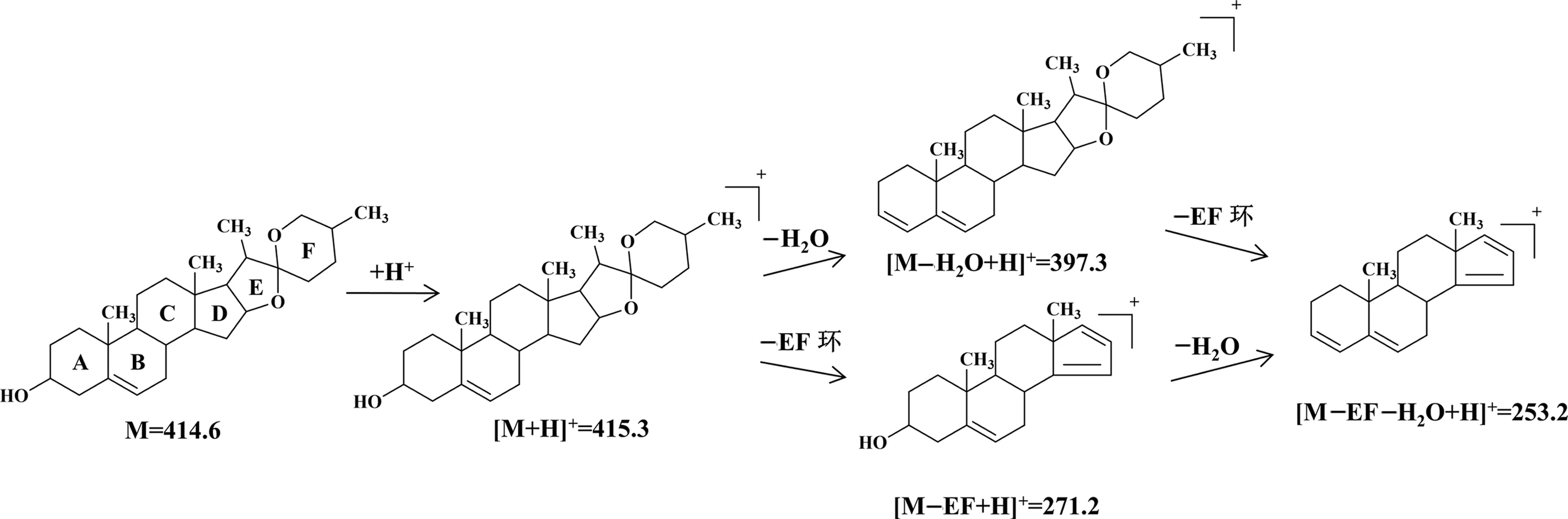
图9 薯蓣皂苷元的潜在裂解规律
4 结论
本文以甲睾酮为内标,建立了快速测定黄精中薯蓣皂苷元的DCBI-MS方法,优化了三角形纸基上样方式、扫描模式、上样体积、DCBI源解吸附温度和放电电压等参数,并对方法的精密度和准确度进行了评估,利用二级质谱图,推测了薯蓣皂苷元潜在的质谱裂解规律。本文建立的DCBI-MS法与传统的HPLC-UV法对比,对实际样品的测定结果无显著性差异,与传统HPLC-UV法相比,DCBI-MS更省时省溶剂。该DCBI-MS法不仅有利于大批量黄精样品中薯蓣皂苷元含量的快速测定和高通量分析,也为其他含薯蓣皂苷元的中药材的分析提供了新的策略。综上所述,DCBI-MS法在复杂基质中药材的高通量分析和快速筛查目标化合物方面具有潜在优势,研究结果可为中药黄精中薯蓣皂苷元的基础研究和质量控制提供参考。
