克里唑替尼及其中间产物的优化合成研究*
2022-12-01刘加艳任宇鹏
刘加艳,任宇鹏
(河南应用技术职业学院,河南 郑州 450042)
肺癌居我国恶性肿瘤发病首位,肺癌按细胞形态可分为小细胞肺癌和非小细胞肺癌(NSCLC),其中小细胞肺癌约占15%,非小细胞肺癌约占85%。肺癌最常见的“驱动突变”基因有3个:KRAS(鼠类肉瘤病毒癌基因)、EGFR(表皮生长因子受体基因)和ALK(间变淋巴瘤激酶基因),在我国的肺癌患者中,3%~8%为ALK突变[1]。
克里唑替尼为口服治疗NSCLC的靶向药物,结构式见图1,该药的靶点为EML4-ALK(棘皮动物微管结合蛋白4与间变淋巴瘤激酶融合基因),在无其他药物治疗的新诊断肺癌患者中,克里唑替尼对74%的ALK突变患者有效,其中84%的患者存活时间超过1年,该药可作为一线药物用于治疗ALK突变的新肺癌患者,也可作为二线药物用于治疗已对传统化疗药产生耐受性的患者[2-3]。

图1 克里唑替尼结构式
1 实 验
1.1 材料与试剂
试剂:3-羟基-5-溴-2-硝基吡啶,上海易恩化学公司;乙二醇二甲醚,上海世通化工;醋酸钯,上海荣立化工;1,1’-双(二苯基膦)二茂铁,上海谱振生物公司;碳酸钾,天津大茂化学公司,均为分析纯。
实验设备:LCMS-8050型液相质谱仪,岛津; 1100型液质联机(LC-MS),安捷伦; ELSD 2000型蒸发光散射检测仪,Alltech;MercurPlus400核磁共振氢谱仪,Varian公司。
1.2 克里唑替尼的合成现状及优化合成方法
克里唑替尼在结构上属于杂环化合物,其结构中包含有苯环?厲吡啶环、吡唑环及含氮六元饱和杂环。其结构基础为:苯环与吡啶环通过醚酯键连接,吡唑环与吡啶环直接相连,吡唑环上的H被含氮六元杂环取代。克里唑替尼的现有方法一般分两个阶段:首先用含苯环化合物与含吡啶的化合物发生酯化反应得到醚片段,再经钯系配合物催化与含吡唑的化合物经Suzuki偶联反应得到终产物。目前的合成方法主要存在以下问题:
(1)合成路线较长,导致收率低;(2)提纯方法多采用柱层析,成本高,操作难度大;(3)合成路线步骤较多,副反应多,产品纯度较低,导致收率低。例如公开号为CN105272966A的专利申请中提供的一种“ALK抑制剂克唑替尼及其类似物或盐的制备方法”,其步骤1和步骤2都采用了柱层析的分离提纯方法,操作复杂,不适于工业化生产。公开号为CN105924431A的专利申请中提供的一种“化合物克唑替尼的合成工艺”,其合成路线包括八个步骤,路线较长,造成后处理繁琐,同样不利于工业化生产[4]。
经优化的克里唑替尼合成路线以3-羟基-5-溴-2-硝基吡啶为起始原料,经Mitsunobu反应后,通过酯化引入吡啶环,再经氨基保护作用,发生Suzuki偶联反应得到终产物7,共四步反应,更适合工业生产,见图2。
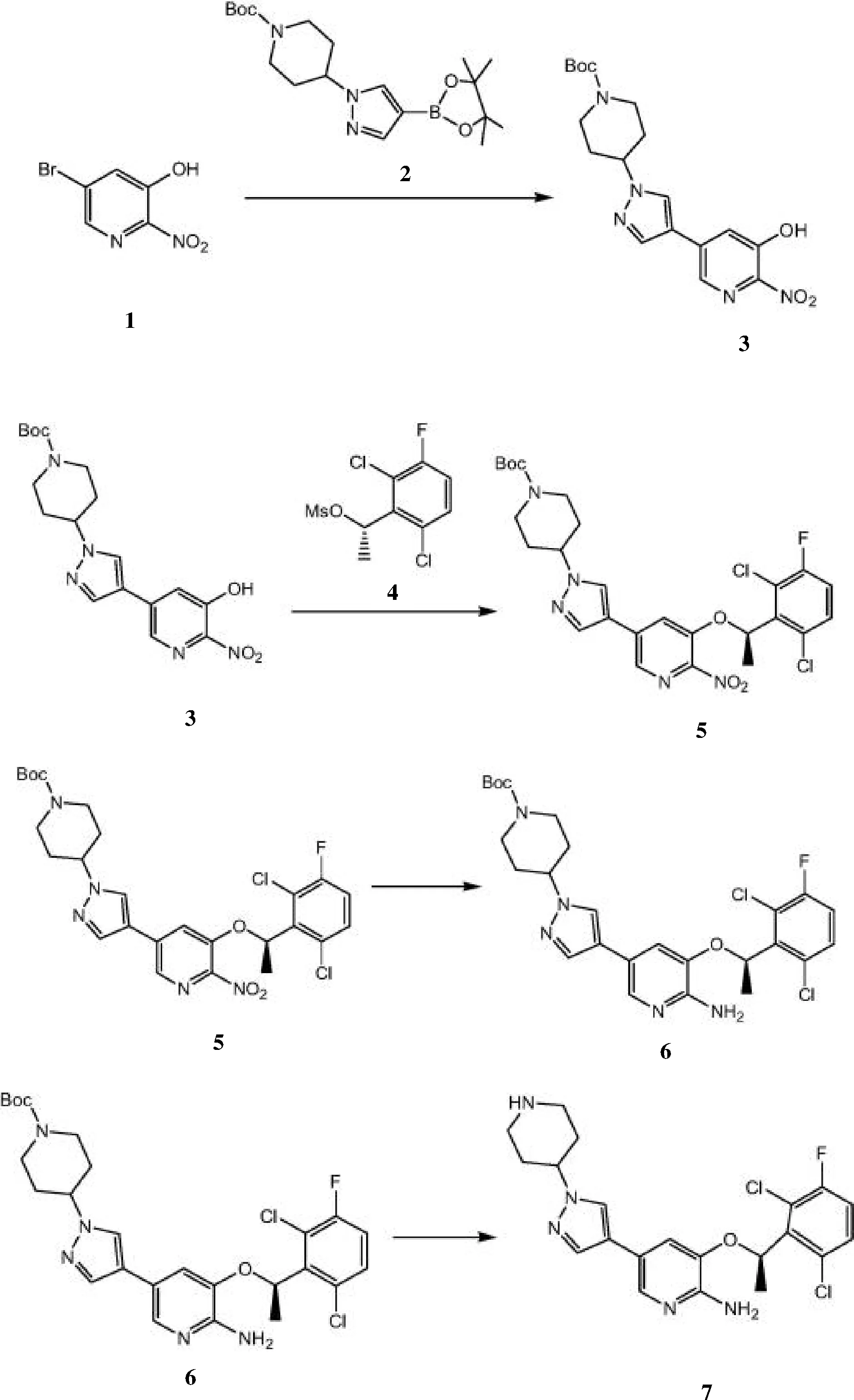
图2 经优化的克里唑替尼合成路线
同时试验以单因素为基础,优化中间产物3,即[1-(1-特戊酰哌啶)-4-(2-硝基-3羟基吡啶-5-基)-1H-吡唑]的合成方法,为中间产物的制备提供参考。
1.3 中间产物3的合成方法
准确量取体积比为2:1的乙二醇二甲醚和水,均匀混合后置于密闭反应容器,加入一定量的醋酸钯和还原铁剂。尽快将一定量的化合物1(5-溴-2-硝基-3-羟基吡啶)和化合物2[1-(1-特戊酰哌啶)-4-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡唑]加入反应容器,以碳酸钾调节反应pH值,加热到90~95 ℃,反应一定时间生成化合物3,即[1-(1-特戊酰哌啶)-4-(4,4,5,5-四甲基-1,3,2-二噁硼烷-2-基)-1H-吡唑]。
生成物为粗产物,提纯处理为:蒸馏溶剂后,向蒸馏残余物加入一定量的水进行抽滤,收集滤液;在滤液中加入乙酸乙酯萃取杂质,收集下层水相;以HCl调节pH至5.5~6.5,即有中间产物3析出,真空干燥后得到化合物3。
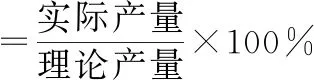
中间产物3得率按下式计算:
2 结果与讨论
以化合物3得率为评价指标,合成试验初始条件为:分别准确称量10.0 g化合物1、19.0 g化合物2及19.0 g K2CO3,迅速加入三口具塞反应瓶中,反应溶剂为乙二醇二甲醚和水的混合液,体积分别比为10 mL和5 mL,在醋酸钯和1,1’-双(二苯基膦)二茂铁催化下进行反应,后续提纯步骤同3所示。 采用单因素试验,研究反应时间、反应温度及结晶pH对中间产物3得率的影响。
2.1 反应温度对中间产物3得率的影响
准确称量反应物、溶剂和催化剂后,准确控制反应温度为:75 ℃、80 ℃、85 ℃、90 ℃、95 ℃、100 ℃,其他条件为:控制反应时间13 h,萃取后的水相用0.1 mol/L的稀盐酸调节pH到6.0,按3所示步骤制备化合物3并测定得率,重复试验3次,取平均值,结果见图3。
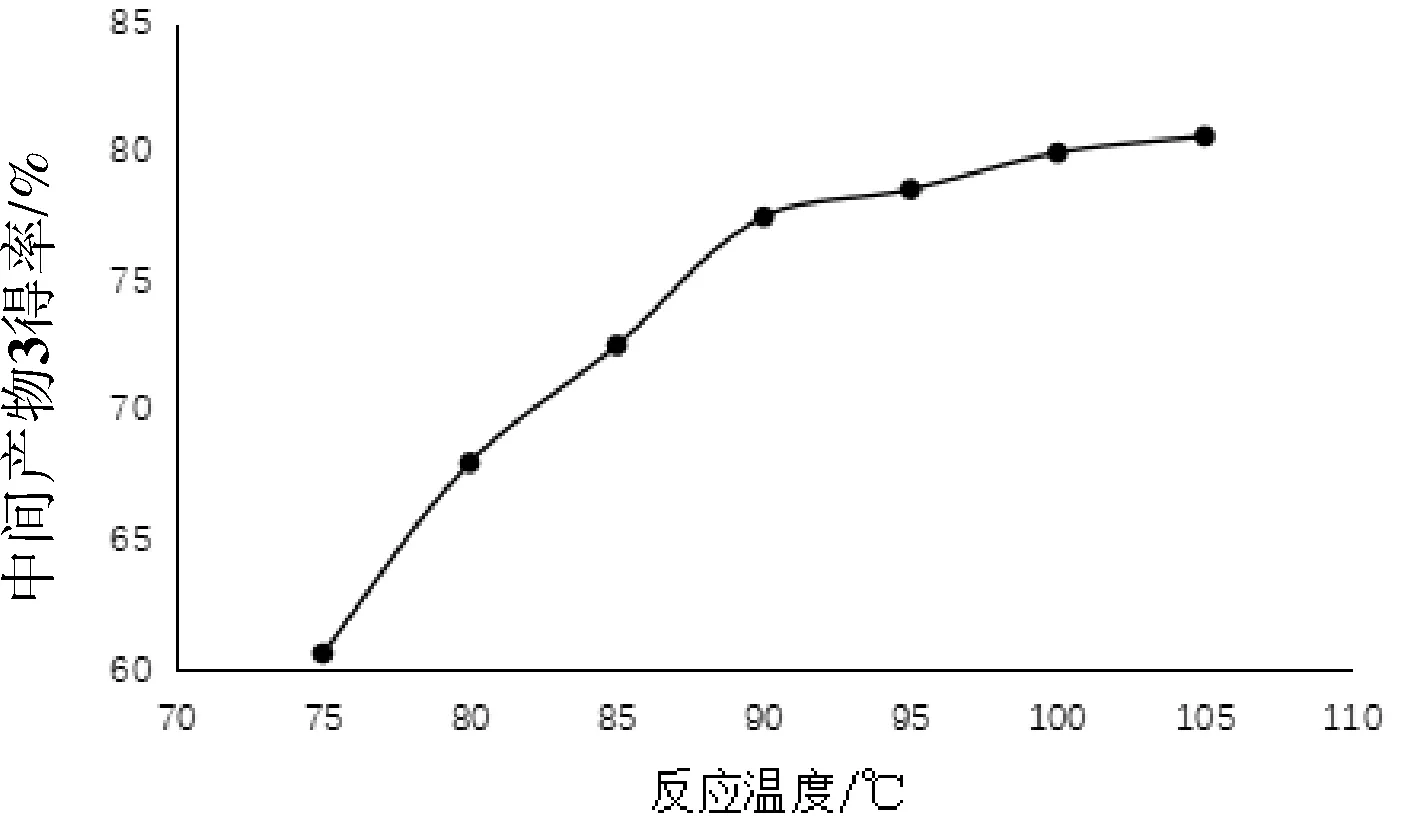
图3 反应温度对中间产物3得率的影响
由图3可知,化合物3得率随着反应温度的增加呈升高趋势,但升高的趋势呈逐渐平缓的趋势,90 ℃以后增速逐渐放缓。温度升高,布朗运动加快,反应速率增加,但温度达90 ℃以上时,副反应也增多,且在后续提纯步骤中,产物3的结晶pH为6.0左右,副产物不在此范围内,因此产率增速逐渐平缓[5]。综合考虑能耗及产物得率,试验设定反应温度为90 ℃。
2.2 反应时间对中间产物3得率的影响
准确称量反应物、溶剂和催化剂后,准确控制反应时间为:10 h、11 h、12 h、13 h、14 h、15 h、16 h,其它条件为控制反应温度为90 ℃,且萃取后的水相用0.1 mol/L的稀盐酸调节pH到6.0,按3所示步骤制备化合物3并测定得率,重复试验3次,取平均值,结果见图4。
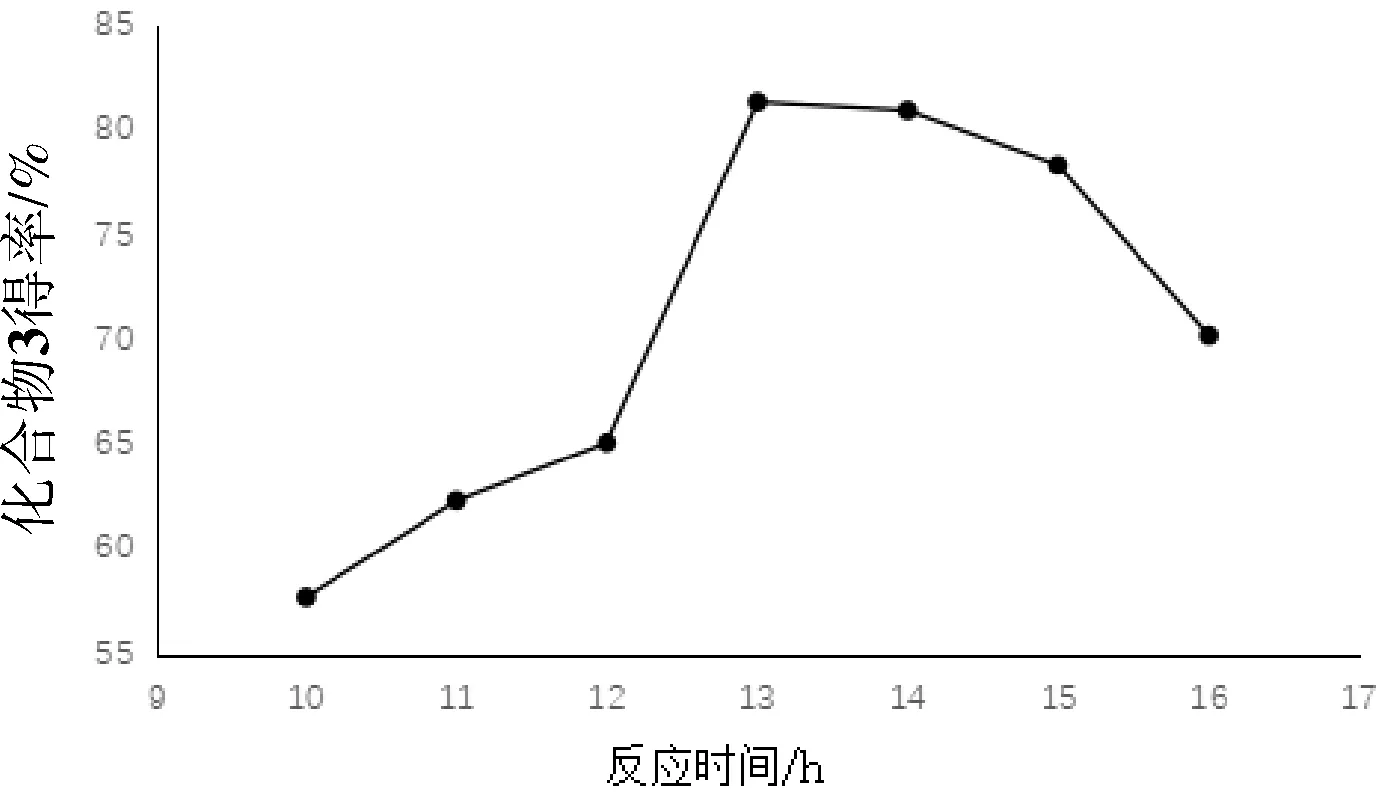
图4 反应时间对中间产物3得率的影响
由图4可知,控制其他条件,随着反应时间的延长,化合物3得率呈现先增加后降低的趋势。其中12~13 h时得率增速最显著,13 h时得率为最大值,13~16 h时得率呈下降趋势。此反应为吡啶衍生物对氢原子的取代反应,为可逆反应,吡啶的化学性质较活泼,随着反应时间的延长,吡啶发生取代或加氢还原,且其他副产物也增加,造成产物得率下降[6],且反应时间长,增加了设备使用周期及热量使用,因此反应时间控制在13 h左右。
2.3 结晶pH对中间产物3得率的影响
按1.3所示步骤制备化合物3,控制反应温度90 ℃,反应时间13 h,反应完毕后经减压蒸馏,萃取抽滤,其中水相滤液用0.1 mol/L的稀盐酸分别调节pH到4.5、5.0、5.5、6.0、6.5、7.0、7.5,并测定得率,重复试验3次,取平均值,结果见图5。

图5 结晶pH对中间产物3得率的影响
由图5可知,随着结晶pH升高,化合物3得率呈先增大后减小的趋势,pH 4.5~5.0产物得率增速最快,pH 5.0~6.5得率趋于稳定在76.34%~80.32%,pH 6.5以上,得率下降。由于产物3具有酸性的酚结构,碱性条件下发生反应,导致副反应增多,得率下降;化合物3又有Boc(叔丁氧羰基)基团,用来脱氨基保护,胺与Boc在弱酸和弱碱(如碳酸钾、三乙胺等)作用才起作用,引起pH过低,副产物也会增多[7-8]。综上,试验中结晶pH控制在5.5左右。
中间产物3的核磁数据及LC-MS检测结果如下:
HNMR(CDCl3,400 MHz) δ=8.51(s,1H),8.26(s,1H),8.18~8.10(m,2H),6.02(s,1H),3.59~3.51(m,1H),3.21~3.19(m,4H),2.15~2.10(m,2H),1.96~1.89(m,2H),1.31(s,9H);LC-MS(M+1):390.2。
3 结 论
从单因素影响考虑,产物3在反应温度90 ℃、反应时间13 h及pH 5.5时得率最高,经实验验证得率为89.2%。经优化的克里唑替尼合成方法,合成需要的步骤少,且每步反应产生的副反应少,提纯简单,不需要进行柱层析分离,实验操作简单,所需设备简单,适用于工业化生产。
