单原子配位结构及与载体相互作用的调控用于二氧化碳电催化还原
2022-11-24陈宇新王丽君姚志波郝磊端谭心怡JustusMasaAlexRobertson孙振宇
陈宇新,王丽君,姚志波,郝磊端,谭心怡,Justus Masa ,Alex W. Robertson ,孙振宇,*
1北京化工大学化学工程学院,有机-无机复合材料国家重点实验室,北京 100029
2 北京理工大学材料科学与工程学院,北京环境科学与工程重点实验室,北京 100081
3 京博哥大学化学系,京博哥,坎帕拉 999123,乌干达
4 华威大学物理系,考文垂 CV4 7AL,英国
1 Introduction
The need for continuous technological progress and development of human society place an ever increasing demand on energy. Fossil fuels are likely to remain the predominant source of energy for the foreseeable future. However, the use of fossil energy will add to the already alarmingly high levels of CO2released into the atmosphere1, which will exacerbate the serious environmental problem of global warming2, leading to continued melting of glaciers, and rising sea levels3. In this regard, the conversion of anthropogenic CO2into valuable products via biochemical4, photochemical5, electrochemical6,thermochemical7and radiochemical reactions8is a noble scientific and technological pursuit. Among these reactions,electrochemical CO2reduction (ECR) is an attractive approach due to its many advantages such as (1) conversion and utilization of renewable energy9; (2) mild reaction conditions; (3)convenient control of the reaction by controlling the applied voltage, as well as catalyst and electrolyte type, among others;(4) production of high value-added chemicals including carbon monoxide, formic acid, acetic acid, hydrocarbons (methane,ethylene, ethane, etc.) and alcohols (methanol, ethanol, npropanol, etc.)10–15. These chemicals can serve as important industrial feedstocks and fuels to reduce our reliance on fossil energy. However, due to the extreme stable nature of the C=O bonds in CO2, a high reaction energy barrier is needed for its activation. In addition, poor product selectivity and the competing hydrogen evolution reaction (HER) under CO2reduction conditions, which impedes the kinetics of CO2reduction, are the key factors restricting the commercial application of ECR16,17. The development of more efficient catalysts for ECR is therefore crucial. Since the ECR proceeds on an electrode surface, and thus the interaction of CO2molecules with the catalyst surface is decisive, it is difficult to study the structure-activity relationship towards the ECR. The local physicochemical properties, pH value as well as catalyst morphology all influence the overall performance18–20. Hence,developing efficient catalysts with well-defined uniform composition and controllable structure is urgently required to improve the effectiveness of the ECR21–23.
Among the leading prospective catalysts for the ECR, single atom catalysts (SACs) have gained increasing attention owing to their peculiar structures and desirable catalytic properties. SACs combine the features of atomically distributed active centers in homogeneous catalysis and good thermal stability in heterogeneous catalysis. Compared with traditional heterogeneous catalysts, the active site structure of SACs is relatively simple, with known electron structure and uniform distribution, which makes SACs a perfect platform to investigate structure-activity relationships of prospective ECR catalysts at the atomic level24–27. From the perspective of ECR, the welldefined active sites in SACs can potentially simplify diverse reaction pathways or weaken the competing HER28–31.Meanwhile, the structural properties of monodisperse sites make SACs more suitable for single proton-electron coupled transfer,but not conducive to the facilitation of C-C coupling reactions,enabling a predominant selectivity for C1compounds14,32,33.Understanding the catalytic mechanisms, and manipulating them via the regulation of SACs from the microstructure level is an important research area for improving catalytic performance18,34. Generally, the active centers of SACs mainly exist as coordination structures, and the catalytic performance primarily depends on the selection of the central metal element,the type and number of surrounding coordination atoms, the nature of the substrates and the modification of the molecules.Moreover, some minor structural modifications can profoundly affect the catalytic performance of SACs19,27,35,36.
At present, there are relatively few reviews on regulating the coordination structure of single atoms and their interaction with the support for ECR. In this contribution, we review strategies for optimizing the coordination structure of SACs for ECR,including selection of central metal elements, heteroatoms coordination, and diatomic metal centers as well as modulation of the surrounding functional groups in the coordination environments of SACs. Finally, we give a perspective on the challenges faced by SACs in the field of ECR and the possible strategies for overcoming them.
2 Fundamentals of single-atom catalysis of ECR
2.1 Thermodynamic process of ECR on SACs
Essentially, the direct one-electron reduction of CO2to generateis the initial step of ECR. This step is however difficult to perform and demands a potential of −1.90 V (vs.standard hydrogen electrode (SHE), in water, pH = 7, Table 1)due to the thermodynamically very stable nature of CO237. The ECR in most cases is proton-dependent. The generation ofcan be avoided, thereby greatly reducing the potential required for the reaction (Table 1). From this scenario, CO2can be converted to different C1products (e.g., CO, HCOOH, CH4, etc.)and C2products (e.g., C2H4, C2H6, C2H5OH, etc.). In SACs electrocatalytic systems, multi-carbon products are difficult to generate due to the atomic dispersion of active sites, and C1compounds are predominantly produced by SACs9. Among them, CO and HCOOH are the two main products.
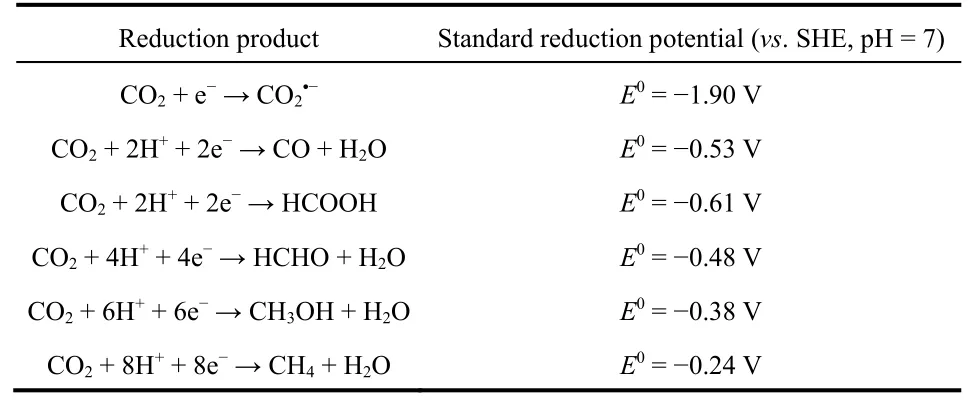
Table 1 Different ECR reactions with corresponding standard reduction potentials.
2.2 ECR reaction mechanisms on SACs
Manipulation of the binding strength of key intermediates is important to attain satisfactory activity and selectivity in the ECR. The formation of *COOH (metallocarboxylic acid)intermediate is considered to be critical for CO production, while the *OCHO intermediate is key for formate generation. Five possible reaction pathways have been put forth for the conversion of CO2to CO on heterogeneous molecular SACs38.Specifically, a CO2molecule can insert into the *M-H (either the metal complex or protonated metal complex) via one electron transfer to generate a metallocarboxylate, followed by furtherreduction and/or protonation to form *COOH. Then CO is desorbed from the metal complex to complete the cycle.Alternatively, the ligand may be involved during the ECR process. The nitrogen in the ligand of a metal complex (or protonated metal complex) is protonated and then initiates the CO2reduction. For formate generation, a CO2molecule is first adsorbed onto a catalyst surface, followed by one-electron reduction to form *and one proton transfer to produce*OCHO. Then formate is released to complete the reaction cycle.
2.3 Suppression of HER on SACs
HER is the major parasitic reaction competing with the ECR.Many studies have shown that SACs present excellent CO2to CO selectivity in aqueous electrolytes, with the competing HER significantly suppressed. This may be related to the isolated nature of active sites in SACs; CO2molecules may preferentially adsorb on the isolated active sites of SACs during the reaction,preventing any H2O interaction, whereas in contrast,nanostructured catalysts with their continuous surfaces can accommodate the adsorption of CO2and H2O molecules simultaneously. Furthermore, since many nanocatalysts are polycrystalline, different exposed crystal planes often exhibit different adsorption capacities for CO2, H2O and reaction intermediates. For example, working electrodes consisting of bulk or nanoparticles of Mn, Fe, Co, and Ni mainly produce H239,while when the metal active sites are dispersed in the form of single atoms, the M-N-C (M = Mn, Fe, Co, Ni) materials are exclusively active catalysts for ECR to CO40.
Density functional theory (DFT) calculations also offer a rational understanding of inhibition of HER on SACs from the energy barrier perspective. Jung et al. compared the initial protonation energy barriers for ECR and HER and found that the energy barriers for the generation of *COOH or *OCHO were lower than that of *H on most SACs (Fig. 1), suggesting that the ECR was more favorable compared to the HER over most SACs28. Bagger et al. suggested that due to the active sites of SACs with a porphyrin-like structure where the *H intermediate tends to bind to the single-atom sites on the top of SACs instead of the hollow sites of metal surfaces, the HER is inclined to proceed through a Volmer-Heyrovsky pathway with less energetic barrier relative to the Volmer-Tafel pathway29.
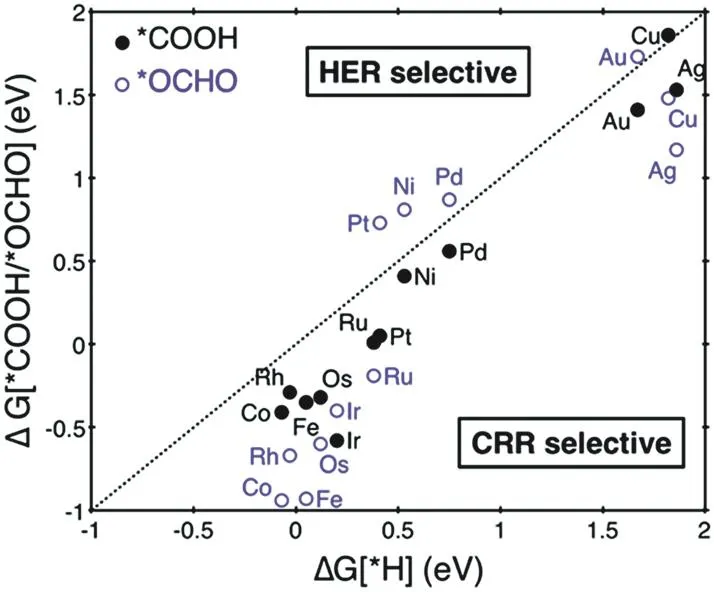
Fig. 1 Free energy change of the first protonation step in ECR and HER on various SACs. Catalysts below the dotted parity line are ECR selective.
3 Types of SACs and their synthesis strategies
3.1 Structure of active sites in SACs
From the thermodynamic viewpoint, a single atom by itself is unlikely to exist alone because of its high surface energy.Anchoring in the active sites of a host material by oxidized state is a common form of SACs. Therefore, selecting a suitable support is indispensable to firmly anchor single atoms by providing strong binding via ionic or covalent interactions.Besides, the electronic state of SACs can be strongly affected by the specific support, thus further influencing the catalytic performance of ECR. A desired support for ECR usually needs to have good electrical conductivity, high specific surface area,sufficient stability, and suitable binding energy with the single atoms41. Thus far, the main supports of SACs for ECR are carbon materials42and metals/oxides43,44.
3.1.1 SACs on carbon-based materials
Carbon-based materials are widely used to immobilize SACs for ECR, which include carbon nanotubes, graphene, and carbon black, etc. Their remarkable electrical conductivity and high specific surface area provide benefits as a SAC support. The most common anchoring method is to introduce some heteroatoms such as M-NxCyVz(M, C, and V denote metal atoms, carbon atoms, and vacancies in support, respectively)45–48.Regulation of the coordination environment permits tuning and optimization of the active sites’ electronic structure for ECR49,50.
3.1.1.1 M-N-C materials
M-N-C represents the most common structure of active sites in SACs for ECR, and has been widely investigated due to their exceptional catalytic efficiency, product selectivity, and stability. These M-N-C materials are usually prepared through bonding between metal atoms in precursors and carbonbased hosts via high-temperature pyrolysis, electrochemical deposition, and physical deposition51. Due to the difficulty in control at the molecular level during fabrication, the coordination environment of central metals in M-N-C catalysts can be complex and heterogeneous52. With the development of more advanced characterization techniques including scanning transmission electron microscopy (STEM),X-ray absorption spectroscopy (XAS), extended X-ray absorption fine structure (EXAFS), among others, the specific structure of individual isolated M-Nxsites can be probed and clarified.
3.1.1.2 Heterogeneous molecular SACs
The immobilization of some molecular catalysts with ECR catalytic activity, such as metal macrocycles (porphyrin,phthalocyanine, cyclam, bipyridine, terpyridine, and phosphine,etc.)53,54or metal macrocycle-based metal-organic frameworks(MOFs), covalent organic frameworks (COFs), and hybrids with chemical stability and structural tunability55–57, onto porous carbon supports with good electrical conductivity is another promising method for the preparation of SACs. These complexes are attached to supports through conjugated grafting58,nonconjugated interactions59or in situ polymerization54.Compared to carbon-based SACs prepared by unpredictable pyrolysis, molecular SACs can be obtained with explicit structures without pyrolysis under harsh conditions. In addition,molecular SACs such as porphyrins and phthalocyanines have well-defined M-N4centers as electrocatalytic active sites, and their electronic structures can be tailored by manipulating the central metal elements and functional moieties60.
3.1.2 SACs on metals/metal oxides
3.1.2.1 SACs on alloys
Metals with high electrical conductivity and electron affinity are another class of promising supports for single atoms. When metal atoms are supported on metal hosts, such SACs often exhibit excellent catalytic performance due to the synergy between the monodispersed metal atoms with the metal support.At present, this type of materials is usually single atoms anchored or dispersed on alloy material or intermetallic nanocrystalline supports, namely a single-atom alloy (SAA).The formation of SAA is achieved resulting from adsorption of metal atoms by the different surface potentials of metal supports61.From the perspective of ECR, the monodispersed sites collaborate with the host metals to provide dual adsorption sites,which may break the linear adsorption free energy constraint of the intermediate *CO and *COOH. For instance, Mueller and co-workers found that Pd-Au alloy (Au nanoparticles decorated with atomically dispersed Pd) achieved a better performance for ECR to CO compared to pure Au, Pd and conventional PdAu alloy catalysts44. Theoretical calculations showed that the Pd-Au alloy balanced the energy barriers of the rate determining step (RDS) of ECR corresponding to pure Au (CO2activation) and pure Pd (*CO desorption), respectively,as shown in Fig. 2. In addition, it is worth noting that for these SAA materials, the strong electrophilicity of metal supports can increase the concentration of electronegative species around the active sites, which could suppress the occurrence of HER and enhance ECR activity.

Fig. 2 Schematic illustration of atomically dispersed Pd sites on Au surface for improved ECR to CO.
3.1.2.2 SACs on metal oxides
Metal oxides are widely utilized as catalyst supports to provide binding sites for single atoms. For example, pentacoordinated Al3+sites have been proposed to immobilize Pt single atoms. In contrast to this electrostatic stabilization, using reducible oxides such as FeOx, TiO2, MnO2, and CeO2enables stabilizing single atoms by forming stronger covalent bonds.Under such circumstances, the binding energy of ionic metal species on these metal oxides rich in defects (e.g., oxygen vacancies) can exceed that of metal atoms on bulk metal. For example, Wang et al. firstly introduced oxygen vacancies on the surface of TiO2by calcination of the metal oxide in an inert atmosphere. Then, the defective TiO2was mixed with the Au precursor solution to form a precipitate under the action of ammonium nitrate. A stable Au/TiO2SAC was generated by further annealing the dried precipitate. The surface defects of TiO2nanosheets were claimed to stabilize Au atoms by creating a Ti-Au-Ti structure62. Using an atomic migration protocol,Wang et al. prepared Ag1/MnO2SACs through reconstruction of Ag nanoparticles on the surface of MnO2(211) to MnO2(310) by high-temperature migration43. Thermal treatment and surface reconstruction of the metal oxide were supposed to contribute to the yield of single Ag atoms. The as-made Ag1/MnO2afforded a FEco of 95.7% at −0.85 V (vs. RHE). DFT computations suggested that Ag single atoms had high electronic density approaching the Fermi level and could serve as the main active sites for the ECR.
3.2 Synthetic strategies of SACs
Synthesis of highly active and robust SACs is still challenging, since discrete single atoms tend to migrate and aggregate/agglomerate into clusters owing to their high surface energy. How to uniformly disperse single atoms on the surface of the host, especially at high metal loadings, and how to fabricate SACs at a large scale remain outstanding problems. We summarize and discuss several typical preparation methods in this subsection.
3.2.1 Heat treatment technologies
3.2.1.1 In situ pyrolysis
SACs anchored on carbon supports are widely produced via a high temperature pyrolysis method. Common precursors for synthesis of SACs using this route include MOFs and their derivatives, graphene, organic polymers, as well as a blend of metal salts, nitrogen and carbon sources, etc. In addition to the ease of operation, altering the pyrolysis temperature enables an easy control of the metal loading, porosity, and coordination structure, thereby regulating the ECR activity of SACs.
MOF materials are composed of metal ion nodes and coordinated organic ligands. In MOFs, the metal ion nodes are far away from each other, and an organic ligand molecule is spaced between the nearest adjacent metal ions. This independent and uniform structure is very beneficial for the synthesis of SACs63,64. Among many pre-designed MOF materials, Zn-containing MOFs are the most suitable precursors for the preparation of SACs by high temperature pyrolysis, due to the lower boiling point of Zn, which can leave more pores in the carbon support by volatilization at high temperature, and thus increases the spacing of central metal atoms to prevent aggregation65. This top-down method has been utilized to synthesize various SACs66,67. Similarly, other SACs synthesis methods, such as molecular cage pyrolysis strategy68and polymer pyrolysis strategy69, have been also developed.
Graphene is a typical carbon support used in the preparation of SACs by pyrolysis. By introducing functional groups and heteroatoms, metal single atoms anchoring sites can be created in graphene, and SACs can be constructed through electrostatic interactions or via heteroatom coordination70,71. Nitrogen doping is a common anchoring strategy in graphene-based SACs72. In a typical preparation process, nitrogen-doped graphene is first prepared by introducing a nitrogen source or a pre-designed nitrogen-containing precursor into the graphene framework, and then the nitrogen-doped graphene and metal source are co-pyrolyzed to obtain graphene-based SACs.Nitrogen-doped graphene has been employed as a support for the synthesis of various SACs (e.g., Fe, Co, Ni, Ru, Mn, and Sc SACs)30,70,71,73.
Pyrolysis of a mixture of metal salts, nitrogen and carbon precursors has been shown to be an effective way to construct SACs with M-N-C configurations74,75. This hybrid pyrolysis method has advantages of flexibility and versatility for synthesis of various single metal atoms. However, the synthesis process lacks controllability of the coordination environment.
3.2.1.2 High-temperature migration
Metal species can form volatile metal oxides or nitrides under high temperature calcination in corresponding oxygen and ammonia atmospheres. The gaseous volatiles migrate with the gas stream and are captured downstream by defect sites in a predesigned support to form SACs76,77. This preparation process requires a higher calcination temperature, and the atmosphere is critical for the formation of metal volatiles. Besides, the concentration of the support defect sites plays a pivotal role in the formation and stabilization of SACs. Transformation from crystalline particles to single atoms could even occur under strong metal-defect interactions78–80. Some metal oxides with lower volatilization temperatures, such as Cu2O, MoO, and SnO,have also been observed to form SACs by high-temperature migration in inert gases81.
3.2.2 Wet chemical method
The wet chemical method does not require special equipment for the preparation of SACs and is convenient to carry out in the laboratory. This type of method is usually carried out in several steps. Firstly, the metal precursors are introduced on the support by means of impregnation, ion exchange, and co-precipitation,etc. under appropriate conditions (pH value, temperature, etc.),followed by drying, then the metal precursors are reduced by calcination, light irradiation, microwave irradiation or other methods. For this preparation method, the strong interaction of metal atoms and the host prevents the migration and aggregation of metal atoms, but the support has a limited ability to enrich metal precursors, so too high a concentration can lead to the formation of metal nanoparticles82–84.
3.2.3 Electrochemical deposition
Compared with traditional thermochemical routes, the electrochemical deposition method is environmentally benign,and typically operates using a facile two- or three-electrode system at ambient temperature and pressure. During the electrochemical deposition process, a bulk metal can be dissolved in solution by adjusting applied potentials followed by re-deposition on the support to form single atoms. For example,deposition of Pt atoms on a support can be accomplished by using a Pt wire. The Pt wire is first oxidized at the anode and dissolves in anolyte, and subsequently is re-deposited on the working electrode85. Note that the metal SACs deposited at the cathode or anode can have different electronic states and mass loading, thus displaying distinct catalytic properties.
3.2.4 Atomic layer deposition (ALD)
ALD is a process in which vaporizable metal precursors are deposited on the support layer by layer by vapor deposition. The ALD technique can utilize continuous, self-limiting surface reactions to precisely manipulate particle size and its distribution of metal species on supports86. A typical ALD process for synthesis of SACs encompasses two steps. Metal precursors first react with adsorbed oxygen on the surface of the support.Subsequently, an oxygen pulse is introduced to oxidize the anchored metal precursor to form metal-oxygen single-atom sites. The shape, size, density, and decoration amount of metal species on the support can be fine-tuned by manipulating the ALD cycles. Graphene and metal oxides including Co3O4, CeO2,and ZrO2have been demonstrated to be suitable supports to prepare SACs by the ALD technology87. However, the metal loading is still low (typically less than 2% by weight) due to the agglomeration of metal species during the repeated ALD cycles.
3.2.5 Ball milling
The ball milling technique applies the kinetic energy generated by ball collisions to grind and mix powder materials,and is often used in heterogeneous catalysis. The temperature and pressure involved during the ball milling process can destroy the molecular precursor and rebuild the chemical bond between metal atoms and support, thereby forming SACs88.
4 Tailored active center elements in SACs for ECR
Different elements have distinct electronic structures, thus differ in catalytic properties for a reaction. Therefore, selection of a suitable central metal element is a prerequisite for constructing efficient SACs for ECR. Fig. 3 shows the common metal elements in SACs, among which transition metals especially Fe, Co, Ni, Cu, and Zn, are the most frequently used because of their low price and earth abundance89. The product selectivity of these SACs for ECR are highly correlated with the binding energies of their active sites to the reaction intermediates, such as *CO, *CO2−, and *COOH. CO is the most common ECR product on these SACs owing to the lack of CC coupling sites on the isolated atoms. Cu and Zn SACs enable deep reduction of the ECR to produce CH4and CH3OH. Formate is the major reduction product for p-block SACs. In this review,we summarize SACs with different central metal atoms, and focus on the underlying causes for the variation in ECR activity of SACs with different central metal atoms from the perspective of the reaction mechanism.
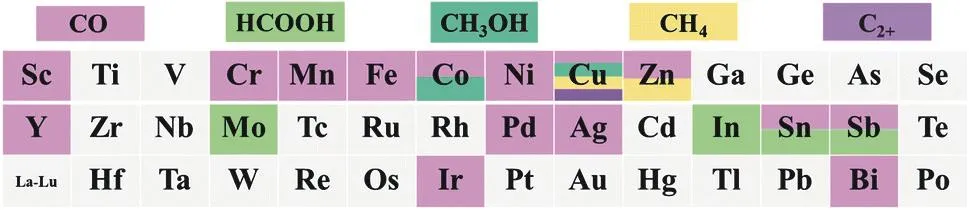
Fig. 3 The most commonly reported central elements in SACs for the ECR.
4.1 d-block metals
A large part of SACs embody d-block metals as the central element, due to the existence of empty d orbitals whose outer electronic structure is easily changed during the reaction. Based on the fact that ECR itself is a multi-step electron-proton pairs coupling reaction, the structural change of the electrons in the outer orbital of the active metal is beneficial to promoting the occurrence of ECR. To date, d-block elements such as Fe, Co,Ni, Mn, Mo, Ru, Rh, Pd, Ir, and Pt have been successfully synthesized and used in ECR90–97. First, from the Pt group noble metals, Pt as a representative has been proved to have a certain ability to catalyze ECR, however, when Pt species exists in the form of conventional clusters, the reaction would be accompanied by a large amount of HER. The application of Pt SACs not only greatly reduced the dosage of precious metals used, but also could effectively inhibit the occurrence of HER.Similarly, noble metals Ir and Ru also showed a certain ability to catalyze ECR whereby the catalytic product was mainly CO98,99. Although these noble metals show certain activity and product selectivity in the application of ECR to CO, their wide application is limited by their high cost. It is therefore essential to develop non-precious metal electrocatalysts. Among them,Ni, Fe, and Co are the three most frequently studied transition metals due to their abundant reserves and excellent catalytic ability, among others.
Ni SACs have been widely studied due to their high CO selectivity. However, bulk Ni-based materials including oxides,hydroxides, metallic nickel, etc., generally show low catalytic performance in ECR100. This is mainly due to the strong adsorption of CO on the Ni(111) face, which poisons the catalyst101,102. Ni SACs were shown to significantly reduce the CO desorption energy, suppress the HER, and enhance catalyst stability103,104. For example, Jiang and co-workers reported a Ni SAC105, using Ni atoms coordinated in a graphene shell as active sites for CO2version, which exhibited a FEco of over 90% with a current density reaching 60 mA·mg−1. Theoretical simulation studies revealed that relative to bulk Ni, the unique electronic structure of Ni single atoms enhanced the conversion of CO2to CO while suppressing HER. A subsequent study examined the effect of four different coordination arrangements of Ni single atoms in the graphite layer on the catalytic performance30. The DFT computations showed that the bulk Ni(111) crystal facet has the highest *CO desorption energy barrier, while the Ni-N moiety with two vacancies as active sites has the lowest CO generation energy. At the same time, the competing HER was very weak on this single atom Ni catalyst owing to large limiting potentials. In Ni SACs, Ni single atoms usually exist in a coordination form with N atoms, where the marked interaction between Ni and N forms the Ni-Nxsites. Yang et al. further investigated the role of Ni-Nxsites for ECR using fine structure characterization106. They synthesized a Ni(I)-N4SAC by anchoring Ni atoms on the graphene. The coordination structure of Ni(I)–N4was revealed by XAS, X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) characterization. The catalyst achieved a FEco of 97% and CO partial current density of 22 mA·cm−1with an overpotential of 0.61 V, meanwhile the FE of 98% was readily maintained when the reaction continued for 100 h. Among them, the electron delocalization phenomenon of the unique 3d9electron configuration in Ni(I) promoted the reaction, while the uncoordinated electrons in the 3d orbital in Ni(I) were transferred to the 2p orbital of C in CO2to transform CO2to, which is the first reaction step in CO2electroreduction.
Fe SACs display comparable ECR performance to that of Ni SACs in terms of CO selectivity. The formed Fe-Nxspecies are often regarded as the active sites. During SACs preparation,single atoms and clusters (and/or nanoparticles) are prone to coexisting, which would impact the ECR catalytic performance to some extent. By mediating the ratio of Fe-N4sites with Fe nanoparticles, the product ratio of CO and H2could be readily adjusted107. Specifically, when the Fe species in the catalyst are exclusively composed of Fe-N4, the catalyst achieved the best FEco, indicating that the Fe-N4sites are the essential active sites for CO generation. The valence state of Fe in the SACs also profoundly affects their catalytic performance. Gu et al. obtained an Fe SAC with the structure of Fe3+-N immobilized on a carbon framework by pyrolysis of Fe-doped ZIF-8 at 900 °C108.Under the preparation conditions of low temperature copyrolysis, Fe was found to exist in the form of atomically dispersed Fe2+. The reaction and simulation results showed that the high valence Fe sites exhibited faster reaction kinetics, lower onset potential, and higher FEco. Noteworthily, it was reported that during the reaction, Fe3+sites would be partially reduced to Fe2+at the applied voltage of −0.50 V (vs. RHE) and the activity would be lost, which again proved the important role of Fe valence on the activity of ECR. In addition, the excessively strong adsorption energy of *CO on Fe-Nxsites is a key factor restricting the activity of Fe SACs. Li and co-workers prepared the FeN4/C catalysts with the Fe-N4structure as the active sites109. The FeN4/C exhibited higher catalytic activity than N-doped carbon and Fe nanosheets, which achieved a FEco of 93% at −0.60 V (vs. RHE). DFT calculations demonstrated that the desorption of *CO was the actual RDS of the reaction, which is similar to the conclusions reached by several other works about Fe SACs for ECR110–112. Therefore, weakening the adsorption energy barrier of *CO is the key to improving the catalytic activity of Fe SACs. In this regard, Pan et al.’s study found that holes constructed on graphene basal planes to support Fe-N4could significantly reduce the adsorption energy of *CO and improved the catalytic activity of ECR to CO (Fig. 4a,b)111. DFT and crystal orbital Hamilton population calculations showed that the Fe-C bond strength formed between Fe-N4sites at the edge of the substrate with *CO was significantly weakened.

Fig. 4 Schematic showing the electrocatalytic CO2 reduction behaviors on (a) pore-deficient graphene bulk-supported Fe-N4 and (b) pore-rich graphene-supported Fe-N4.
Compared with Ni and Fe SACs, there are relatively few reports on Co SACs in ECR, which may be due to the relatively weak reactivity of Co SACs. But some Co SACs also exhibit unique structures and properties. Hou et al. synthesized a Co SAC (Co-Tpy-C) with a well-defined Co-N4-C structure by pyrolysis of a Co polypyridine complex as precursor113. Co-Tpy-C has an extremely low Co loading (0.011% (w)) and exhibited better catalytic activity than Co doped carbon and N-doped carbon in activity tests, which achieved a FEco of 98% at−0.80 V (vs. RHE) and maintained unattenuated performance with the FE steadily above 80% for 24 h. DFT calculations indicated that the formation of *COOH is the RDS of the reaction, which is also consistent with several other works on Co SACs114,115. Tuning the number of atoms coordinated to the Co single atoms is an effective means to accelerate the formation of*COOH. Wang et al. found that by increasing the pyrolysis temperature, reducing the coordination number of N around the Co sites and forming unsaturated Co-N2sites were beneficial to the activation of CO2116. Compared with the saturated Co-N4, the catalytic activity of Co-N2was significantly improved,reaching a FEco of 94% at an overpotential of 0.52 V.Mechanistic analysis showed that a lower N coordination number led to more unoccupied 3d orbitals of Co atoms, which was beneficial to the adsorption ofand enhanced the reduction rate of CO2. The main reduction product of Co SACs for ECR is CO, and some studies have shown that Co SACs also have a certain ability to afford a deep reduction of CO2. Wu et al. found that the binding energy between Co-N4with CO is moderate compared to Fe-N4and Ni-N4117. To explore possible Co-based sites for CO2deep reduction, they synthesized a CoPc/CNT catalyst using a non-covalent trapping strategy with Co phthalocyanine supported on carbon nanotube. The high electrical conductivity of individually dispersed CoPc and CNTs significantly enhanced the ability to catalyze the deep reduction of CO2. CoPc/CNT achieved a CH3OH FE of 44% at −0.82 V(vs. RHE) with a partial current density exceeding 10 mA·cm−2in 0.1 mol·L−1KHCO3solutions.
4.2 ds-block metals
As a unique metal, Cu has been extensively reported in the field of ECR due to its moderate adsorption energy with CO2reduction intermediates and the capability to promote C-C coupling for C2+compounds production118–122. However,distinctive from bulk Cu-based catalysts, the main catalytic product of Cu SACs was C1, because the distance between single Cu atoms in SACs is large, which is not conducive for the occurrence of C-C coupling reactions123. Formation of the final products of ECR depends on environmental factors around the Cu sites, such as the Cu loading, the coordinated N configuration, and the actual coordination environment. Yang et al. modified porous carbon nanofibers with Cu single atoms to obtain Cu SAs/TCNFs for ECR124. EXAFS spectroscopy revealed the Cu-N4structure of the catalyst. The catalyst exhibited 44% FE for CH3OH formation. DFT computations showed that the Cu-N4sites had a higher binding energy for*CO and the energy barrier for further conversion of the intermediate *COH to *CHOH at the Cu-N4sites was 0.86 eV,much lower than the energy barrier for the conversion to *C.Hence the ECR product was CH3OH instead of CH4. By manipulating the pyrolysis temperature of Cu-based MOF precursors, the content of Cu atoms or Cu-Nxsites anchored on N-doped carbon could be regulated, thereby mediating the ECR product selectivity125. The Cu-N-C-800 obtained by pyrolysis at 800 °C had a Cu loading of 4.9% (molar fraction).The resultant high concentration of Cu-N2sites was observed to be conducive to C-C coupling, which achieved C2H4as the main reduction product with a FE of 24.8% at −1.4 V (vs.RHE, Fig. 5a). When the pyrolysis temperature was elevated to 900 °C, the Cu content decreased to 2.4% (molar fraction). The resulting isolated Cu-N4sites and Cu-N2sites were seen to favor the formation of C1products, affording CH4as the major reduction product with a FE of 38.6% at −1.6 V (vs. RHE, Fig.5b). Karapinar and co-workers prepared a Cu-N-C catalyst with Cu-N4sites by ball-milling followed by high temperature carbonization126. The catalyst produced CH3CH2OH with a FE of 55% at −1.6 V (vs. RHE) in 0.1 mol·L−1CsHCO3. It was found that isolated Cu atoms were instantaneously transformed into Cu nanoparticles during the ECR, but the material reverted to Cu single atoms after electrolysis, which was further verified by operando XAS characterization (Fig. 5c). This intermediate state of Cu nanoparticles formed during electrolysis may be the real source of catalytic activity. An analogous situation was also observed by Xu and co-workers, where Cu single atom sites formed Cun(n = 3, 4) clusters during the reduction process127,and after the electrolysis process, the Cunclusters were oxidized to the original single atom structure. The catalyst achieved the conversion of CO2to CH3CH2OH with a FE of 91% at −0.70 V(vs. RHE), and also exhibited stability for at least 16 h at −0.70 V. This phenomenon of reversible conversion of active sites is beneficial to our understanding of the real active sites of Cu SACs during catalytic process. At the same time, the ability of Cu to deeply reduce CO2cannot be ignored either. Taking Cu atoms as the core to achieve deep reduction of CO2by regulating the surrounding coordination environment is an important research direction.
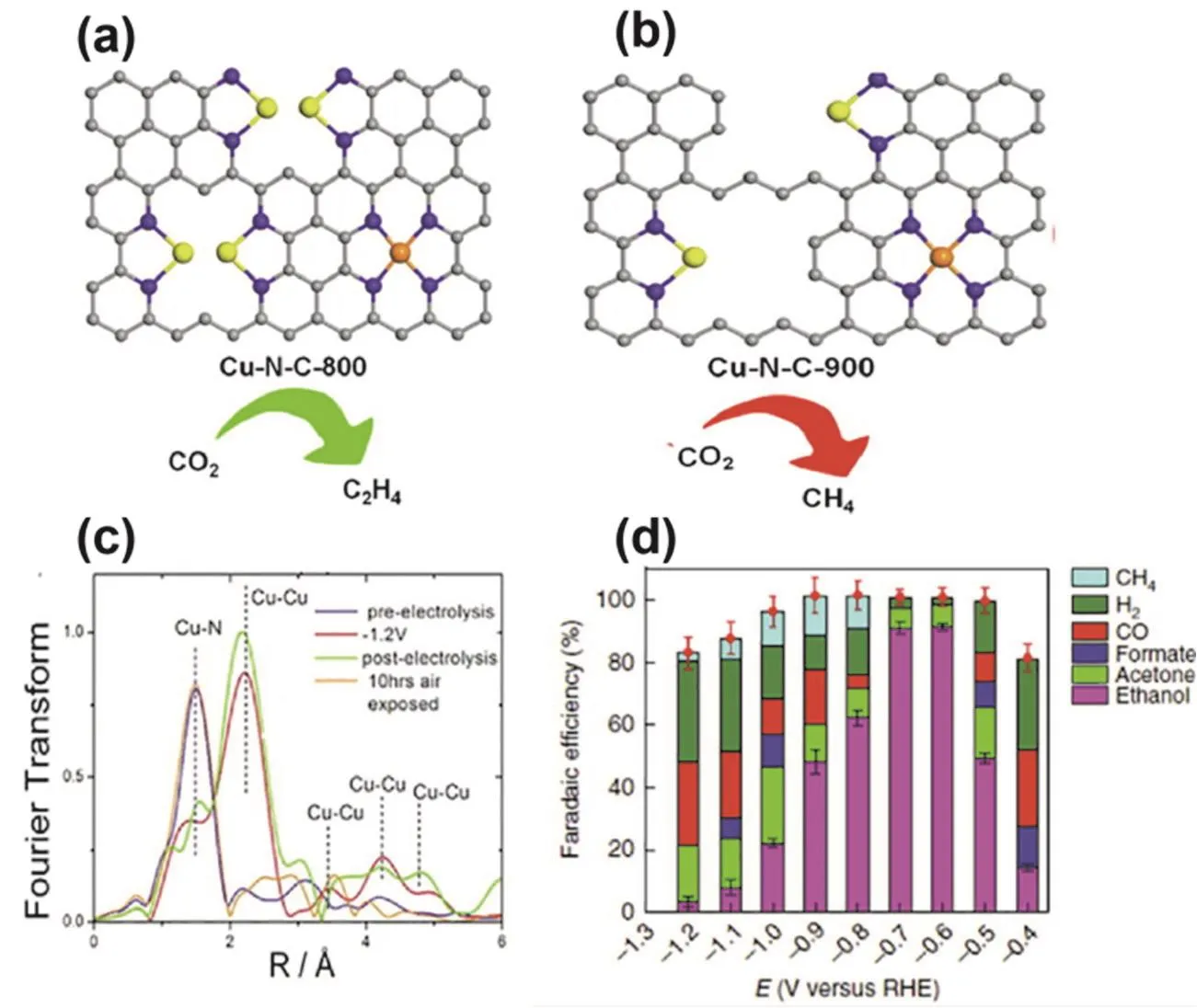
Fig. 5 (a) Cu-N-C-800 and (b) Cu-N-C-900 and their ECR catalytic trends. (c) Operando EXAFS spectra of Cu0.5NC under no potential applied (blue line), during electrolysis at −1.2 V (vs. RHE) (red line), after electrolysis under no potential applied (green line) and after electrolysis at−1.2 V (vs. RHE) then sample exposed to air for 10 h (orange line). (d) FE and the product distribution at different polarization potentials.
The content of Zn in Zn SACs is usually low, because Zn has a relatively low boiling temperature (907 °C), which also leads to the volatilization of Zn when the organic ligands are carbonized. The application of Zn single atoms in the field of SACs is mainly divided into two categories, as the catalysts to generate CO128,129or CH432,130; or only as an additive for optimizing SACs131. Han and co-workers reported a Zn/MNC SAC (Zn-N4embedded in microporous N-doped carbon)32,which gave a CH4FE of 85% at −1.8 V (vs. RHE). The FE maintained at 84% with a current density of 39.9 mA·cm−2after 35 h of continuous polarization. The generation of *CHO was found to be the RDS for Zn/MNC SAC in ECR to CH4.
4.3 p-block metals
p-block metal (e.g., In, Sn, Sb, and Bi)-based SACs have attracted research interest due to their low overpotential for ECR and high selectivity for HCOOH or HCOO−products. Similar to transition metal SACs, M-Nxis also the common structure of the active sites for p-block metal SACs. For instance, Shang and co-workers reported an In SAC with a structure of Inδ+-N by encapsulating In(acac)3with ZIF-8 followed by high temperature carbonization33. EXAFS showed that there was no In―In bond, instead, the first coordination shell of the In atom was comprised of four N atoms, and the length of the In-N bond was 1.6 Å, which is consistent with the In-N4structure.The catalyst exhibited high selectivity to formic acid, achieving a FEHCOOHof 96% at −0.95 V (vs. RHE), and the turnover frequency (TOF) reached 12500 h−1. Xie and co-workers anchored Sn atoms on N-doped graphene to obtain a Sn SAC with a Snδ+-C2N2structure132. The charge density of Sn atoms transferred to the graphene due to the bonding interaction, thus forming positively charged Snδ+centers. The catalyst achieved a FEHCOOHof 74.3% at −1.6 V (vs. RHE), and the TOF reached 11930 h−1. In situ Fourier transform infrared spectroscopy(FTIR) showed that Snδ+sites stabilizedand HCOO−intermediates. Jiang et al. constructed a Sb SAC by anchoring the Sb―N4sites on N-doped carbon nanosheets133. They found that the yield of HCOO* through the first concerted protonelectron transfer was more favorable than *COOH formation according to DFT calculations, so the main product was formate rather than CO. Distinct from In, Sn, and Sb, Bi SACs are more inclined to reduce CO2to CO rather than formic acid or formate.Zhang et al. obtained a Bi SAC (Bi-SAs/NCs) by pyrolysis of Bi-MOFs64. The structure of active sites was Bi-N4with a valence state of Bi falling between 0 and +3. The Bi-SAs/NC achieved a FECOof 97% at −0.50 V (vs. RHE), outperforming Bi clusters and Bi nanoparticles.
5 Regulation of SACs’ microenvironment for ECR
In addition to the composition of the central metal itself, the local environment of active centers and properties of substrates also affect the catalytic activity of SACs for ECR. This section summarizes some methods for regulating the microenvironment of SACs, including heteroatoms coordination, diatomic metal centers, adjusting the substrates, and the effect of surrounding functional groups for SACs.
5.1 Heteroatom coordination
5.1.1 Tailoring nitrogen atom coordination number
Generally, in SACs, the M-N4is the most typical coordination configuration of the metal active center, but this coordination configuration does not necessarily always render good catalytic activity. Regulating the number of coordination atoms around the central metal atom is one of the most direct and effective ways to tailor the properties of the active sites. The most common way to prepare SACs with a lower coordination number is high-temperature pyrolysis. For example, Sa and coworkers applied heat treatment to graft phthalocyanine molecules on carbon nanotubes and obtained a NiPc/CNT catalyst with the Ni2+-N4structure134. X-ray absorption fine structural characterization showed that the coordination environment of Ni species in the calcined NiPc/CNT was gradually transformed into Ni+-N3V (V represents vacancy)and Ni+-N3. Among them, the coordination number of the Ni center was reduced from 4 to 3, and the charge was also lowered from +2 to +1, respectively. ECR tests showed that the calcined NiPc/CNT with Ni+-N3sites delivered a superior catalytic activity for CO2to CO compared to the NiPc/CNT with Ni2+-N4.DFT computations indicated that the energy barrier for the generation of *COOH on the Ni+-N3sites was much lower than on Ni2+-N4ultimately leading to the observed difference in catalytic activity. Similarly, Gong et al. synthesized a series of Ni SACs with varying N coordination numbers (NiSA-Nx-C)135.The ECR results showed that the low-coordinated NiSA-N2-C with Ni-N2sites exhibited much higher CO FE (98%) and TOF(1622 h−1) than that of NiSA-N3-C and NiSA-N4-C. The X-ray absorption fine structure characterization of the catalysts is shown in Fig. 6a–d. DFT calculations indicated that decrease in N coordination number promoted the formation of the*COOH intermediate in the Ni-N2sites to increase the catalytic activity of ECR. The optimization strategy was based on lowering the coordination number of N with the center metals,which was also attributed to Cu and Co SACs for improving the ECR to CO activity116,136. For instance, Feng and co-workers demonstrated that the Cu-N2/GN nanosheet catalyst with a low N coordination number provided a FECOof 81% at a low potential of −0.50 V (vs. RHE)136, superior to Cu-N4/GN. DFT calculations revealed that the Cu-N bond length was shorter in Cu-N2than in Cu-N4, which is expected to be beneficial for electron migration from Cu-N2sites to *CO2, further promoting the formation of *COOH and *CO intermediates.
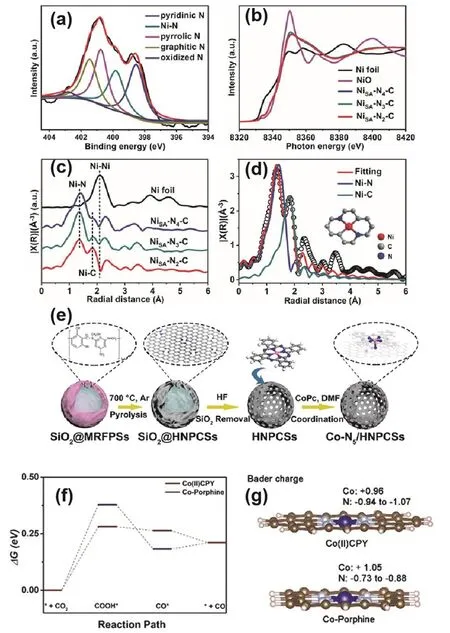
Fig. 6 (a) XPS spectrum of N 1s for NiSA-N2-C. (b) Normalized Ni K-edge XANES spectra and (c) FT-EXAFS spectra of NiSA-Nx-C and Ni foil. (d) EXAFS fitting and optimized model for NiSA-N2-C. (e) Schematic illustration of Co-N5/HNPCSs. (f) Free energy profiles of Co(II)CPY/graphene and Co-porphine/graphene. (g) Bader charge of Co and N atoms.
Introducing additional N sources in the vicinity of M-Nxsites can increase the N coordination numbers. This strategy is conducive to inducing axial coordination between the center metal and the extrinsic N source. An Fe SAC with Fe-N5sites was first reported by Cheng and co-workers. The composite catalysts were constructed by encapsulating FexN nanoparticles in Fe-N5sites embedded in graphite sheets by means of electrospinning, carbonization in an inert atmosphere137, and nitridation in an ammonia atmosphere. The catalyst achieved a FEco of 95% and partial current density of 4.71 mA·cm−2at−0.53 V (vs. RHE). The Fe-N5sites, among others, were obtained by co-pyrolysis of heme, melamine and graphene,followed by acid leaching. Before adding graphene, only an Fe-N4structure was obtained. During the pyrolysis of melamine, N heteroatoms were doped into the introduced graphene, and the Fe-N4sites dispersed on the support were axially coordinated with the doped extra N atoms, resulting in Fe-N5sites. DFT calculations indicated that the additional axial coordination led to further reduction of electrons in the Fe 3d orbital, thereby reducing the feedback π bond between Fe and CO, in effect promoting CO desorption. The catalytic activity of the Co-Nxstructure can also be enhanced by introducing additional N sources. Pan et al. prepared nitrogen-doped carbon spheres with a hollow structure using a sacrificial template method138, which adsorbed cobalt phthalocyanine (CoPc) via impregnation to form Co-N5/polymer-derived hollow N-doped porous carbon spheres (HNPCSs) with a Co-N5structure (Fig.6e). The catalyst maintained a FECOof above 90% under a wide potential window (−0.57 to −0.88 V). Both current density and FE were preserved after 10 h of continuous catalysis. Theoretical calculations showed that Co-N5possessed a lower free energy to form *COOH and desorbed CO more easily than Co-N5−xsites.
5.1.2 Tuning nitrogen species configurations
Nitrogen species exist in SACs in the form of pyridine-N,pyrrole-N, graphite-, and oxide-N. Metal active centers usually form bonds with pyrrole or pyridine-N. The d-band center of pyrrolic-N coordinated metal sites is reported to be lower than that in pyridine-N coordinated metal sites, imparting a relatively weak adsorption strength of the intermediate transition state.Wang et al. constructed two types of Co SACs, i.e., Co(II)CPY(Co atoms bonded with four pyridinic N atoms) and Co-porphine(Co atoms bonded with four pyrrolic N atoms)139. Co(II)CPY was observed to have better ECR activity than Co-porphine.DFT calculations revealed that the first electron transfer to form the *COOH intermediate was the RDS. Co(II)CPY was calculated to possess a lower reaction energy barrier for this step(Fig. 6f). Bader charge investigation further indicated that the Co atoms in Co(II)CPY obtained more electrons from the pyridine nitrogen (Fig. 6g). Projected density of states revealed that the dz2orbital in Co(II)CPY was closer to the Fermi level prior to the ECR reaction, while there was an overlap with the 2p orbital of*COOH after the reaction. This supported that the adsorption of*COOH on Co(II)CPY was enhanced.
However, in the study of Hu et al., the coordination strength of pyrrolic nitrogen was higher than that of pyridine nitrogen.They prepared Fe SACs with two configurations of pyrrolic nitrogen (Fe3+-N-C) and pyridine nitrogen (Fe2+-N-C),using different pyrolysis temperatures. Fe3+-N-C afforded better catalytic performance, attaining a FECOof 95% and partial current density of 94 mA·cm−2with an overpotential of 0.34 V.Operando XAS demonstrated that the Fe species in the pyrrole nitrogen coordination configuration existed in the form of Fe3+ions, which maintained stability during the catalytic process.Relative to Fe2+-N-C, Fe3+-N-C was observed to impart more advantages in CO2activation and CO desorption. There remains a controversy about the preferred N functionalities for ECR, which warrants further exploration for different SACs.
5.1.3 Coordination with other heteroatoms (besides N)
Heteroatom doping not only impacts the electron transfer properties of the central metal element but also induces defects on the substrate, thereby modifying the electronic structure and catalytic performance of SACs. The electron-rich properties of oxygen and sulfur make them suitable dopants to be introduced into the coordination environment of SACs. As an example,Wang and co-workers synthesized an Fe SAC supported on porous nanocarbon with an Fe-N4O structure140. In the Fe-N4O, the four nitrogen atoms were coordinated with the Fe atom in-plane, while the introduced O was axially coordinated to the Fe. DFT calculations revealed that the Fe-N4O sites greatly reduced the desorption energy of CO, making the reaction RDS alter from *CO-to-CO to CO2-to-*COOH, thus facilitating good reactivity at relatively lower overpotentials. The Fe-N4O catalyst provided a FECOof 95% at −0.57 V (vs. RHE). In another work, Yang and co-workers prepared two types of Ni SACs anchored on N, S-doped graphene (A-Ni-NSG) and N-doped graphene (A-Ni-NG), respectively106. The XRD diffraction peaks of graphite became significantly weakened after S doping, indicating the formation of defect sites. Multiple characterizations demonstrated the formation of corresponding Ni-S bonds in A-Ni-NSG. In the electrochemical tests, ANi-NSG delivered larger current density, lower reaction overpotential and better stability, suggesting that S doping substantially improved the ECR catalytic performance. In addition to O and S, doping with the more electronegative Cl and F elements has also been shown to effectively improve the ECR catalytic activity. Generally, Mn SACs indicated no promise of good product selectivity when applied in ECR due to their strong adsorption of CO. Zhang and co-workers found that addition of Cl into Mn SAC ((Cl, N)-Mn/G) could markedly improve the ECR selectivity141. Notably, the CO selectivity was improved from 17% to 97%, and the CO partial current density was increased by 11 times. Partial density of states (PDOS) revealed that after doping with Cl, the d-band center of (Cl, N)-Mn/G was lowered compared to that of MnN4, thereby lessening the CO adsorption energy and enhancing the ECR selectivity. DFT calculations further provided supporting aspects, with the energy barrier of the RDS reducing from 1.64 eV for MnN4to 0.65 eV for (Cl, N)-Mn/G (Fig. 7a). At the same time, the energy barrier of HER increased from 0.11 eV for MnN4to 0.92 eV for (Cl, N)-Mn/G, signifying that HER was strikingly inhibited (Fig. 7b).Adjusting the doping heteroatoms can also affect the reaction pathway of ECR, resulting in different final products. For example, Ni et al. embedded Sn-Oxmononuclear species in carbonized ZIF-8 by chemical vapor deposition142, followed by fluorination via etching ZIF-8 with C2F4generated by the pyrolysis of tetrafluoroethylene to form Sn SAC with a Sn-C2O2F2structure. The catalyst displayed a FECOof 90% over a wide voltage range from −0.20 to −0.60 V (vs. RHE). It is worth noting that under the same electrocatalytic conditions, Sn-N4yielded formate as the main product, while Sn-C2O2F2facilitated optimized adsorption of the intermediates through modulation of the coordination structure by the C and O atoms,as well as the axially coordinated F atom, inhibiting the HER and promoting the conversion of CO2to CO.
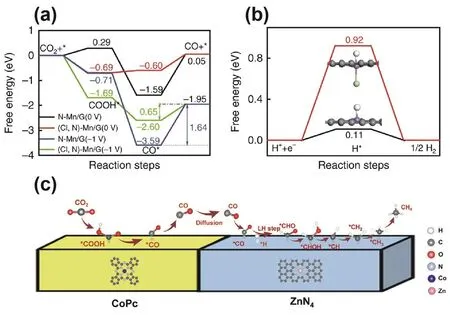
Fig. 7 Calculated free energy of (a) the ECR and (b) hydrogen adsorption.(c) A proposed reaction mechanism of ECR to CH4 over CoPc@Zn-N-C.
5.2 Construction of dual sites
5.2.1 Heteronuclear bimetallic atoms
Because ECR involves the transfer of multiple groups of electrons and proton pairs, SACs with single active sites, low loading and long distance between the key active site atoms, are not conducive to facilitating the occurrence of multi-electron reactions. Single-atom alloys composed of heteronuclear bimetals can generate synergistic effects, which favors the formation of C2+products or improves the conversion efficiency of C1products143,144. In SAAs, the individual active sites are usually dispersed to avoid bonding between adjacent metal sites145. Theoretical calculations can be used to predict the catalytic performance of SAAs for ECR. For example, according to the differences in carbonphilicity and oxophilicity among various elements, Wang et al. simulated 21 different models of heteronuclear bimetals supported on monolayer C2N(M1M2/C2N) and compared their performances in ECR146.Based on the adsorption relationship of the reaction intermediates *COOH, *CO, and *CHO on these diatomic sites,CuCr/C2N, CuMn/C2N, FeCr/C2N, and FeMn/C2N were considered to be promising for ECR, while CuCr/C2N and CuMn/C2N exhibited an interesting potential for deep conversion of CO2to CH4at a more positive onset potential.Regulation of heteronuclear bimetallic SAAs allows for enhanced ECR activity. Li et al. designed two diatomic catalysts NiMn-C3N4-CNT and the NiCu-C3N4-CNT and compared their ECR with the corresponding mononuclear catalysts ((Ni, Cu, Mn)-C3N4-CNT)147. The diatomic catalysts provided higher catalytic activity, imparting a FEco of 90% over a potential window from −0.6 to −0.9 V (vs. RHE).XPS results indicated that Ni, Cu and Mn were all at a higher valence state, indicating that their electronic structures were affected by the doping of the second metal. The M-N bond length in bimetallic sites was calculated to be shorter than that in single atom sites. These results implied that the interaction between diatoms promoted the ECR catalytic performance.Doping the Cu-based support with another single metal atom to prepare SAAs provides a way to deeply reduce CO2by tuning the adsorption energy of the reduced intermediate state. For instance, Zhong and co-workers developed a machine learning approach to explore the catalytic capabilities of 244 different Cucontaining intermetallic crystals148. The resulting volcano plots of catalytic activity and selectivity showed that when the CO binding energy was around −0.67 eV and the H binding energy was above −0.20 eV, the process of CO2reduction would predominate over the HER. Further t-distributed stochastic neighbor embedding (t-SNE) diagram showed that the Cu-Al alloy possessed abundant adsorption sites and suitable ∆ECOvalues with the Cu-Al bridge structure on the Cu surface being the most active. Theoretical calculations also predicted that the Cu-Al sites had promising potential for CO2reduction.According to theoretical calculations (about 10% Al on the surface), a Cu-Al catalyst was fabricated, which gave a C2H4FE above 80% with a partial current density of 400 mA·cm−2and a cathodic C2H4energy conversion efficiency of 55% ± 2% at 150 mA·cm−2.
Tandem bimetallic centers afford a synergy that can break the scaling relationship of the reduction intermediate adsorption energy, improving the catalytic efficiency and favoring deep reduction149. Using phthalocyanine molecules as organic ligands to coordinate with metal junctions provides a common way to prepare tandem electrocatalysts. For instance, Lin and coworkers reported a tandem catalyst comprising CoPc and Zn-N-C (CoPc@Zn-N-C) that can catalyze the ECR into CH4130.The reduction reaction proceeded via two steps. CO2was initially reduced to CO on CoPc, which migrated to adjacent Zn-N4sites for further reduction to CH4(Fig. 7c). DFT computations showed that the activation potential of the N atoms in CoPc to hydrogen also had a synergistic effect on the subsequent further reduction of CO to CH4on Zn-N4sites. A predominant number of SACs have been shown to display remarkable catalytic ability for ECR to CO, while Cu-based materials have the potential to deeply reduce CO2due to their moderate intermediate adsorption strength. Therefore, it is reasonable to combine SACs and Cu-based materials to enable enhanced deep CO2reduction. In this regard, Wang et al.reported an efficient Cu/Ni-NC tandem catalyst for ECR, using a CO/CO2gas mixture as the reactant150. Experiments showed that Cu/Ni-NC had separate adsorption sites for CO2and CO.Ni-NC SACs with large specific surface area acted as an active site for efficient CO formation, while CuOxnanosheets were responsible for catalyzing the C-C coupling reaction. The tandem CuOx/Ni-NC (1 : 4) catalyst has a similar C2H4production rate as the individual CuOxcatalyst with a two-fold mass loading. CuOx/Ni-NC (1 : 2) had the best catalytic performance for ECR to C2H4.
5.2.2 Homonuclear dual-sites SACs
A Cu-based homonuclear SAA was prepared by immobilizing Cu single-atom pairs on a Pd10Te3alloy. XAFS and HAADFSTEM characterization indicated that the Cu species existed in the form of Cu0-Cux+, with the catalyst achieving a FEco of 92% at −0.78 V (vs. RHE)151. DFT calculations suggested that Cu0tended to adsorb CO2molecules while Cux+tended to adsorb H2O molecules, which together promoted the activation of CO2molecules.
5.3 Engineering of substrates
5.3.1 Defect regulation
In addition to direct regulation of the coordination environment around the central atom, creating defects in the substrate can also affect the electron distribution around the single atom active sites and indirectly tailor the interaction between the active sites with the reduction intermediates152. As an example of using metal oxide as a substrate, Zheng et al.synthesized Cu SACs by anchoring Cu single atoms on CeO2with rich oxygen vacancies for boosting CO2activation to produce CH4153. EXAFS spectra indicated that Cu species existed in the form of single atoms when present in CeO2with a concentration less than 5%. When the concentration of Cu was 4%, the coordination number of Cu-CeO2-4% was about 5,suggesting 3 oxygen vacancies around a substituted Cu atom on CeO2surfaces. Theoretical calculations showed that CuCex−1O2x−3possessed the highest CO2adsorption energy and the highest CH4FE (58%) when three oxygen vacancies existed on the surface of CeO2(111) (Fig. 8a,b). Similarly, Guo et al.demonstrated that the synergy between Sn single-atom sites154,oxygen vacancies, and the CuO substrate enabled higher electric double-layer capacitance, superior CO2adsorption capacity, and lower interfacial charge transfer resistance. Experiments combined with theoretical calculations revealed that Sn1/VOCuO-90 promoted the formation of *CO by lowering the*COOH dissociation energy barrier. The formed *CO was adsorbed onto the CuO sites and further reduced to CH3OH. The catalyst imparted a CH3OH FE of 88.6% at a current density of 67 mA·cm−2. There are also several studies reporting that defects in carbon substrates can greatly impact the catalytic activity155–157.For example, Li et al. attached Fe-N4sites to defective nanoporous graphene oxide using a H2O2etching strategy111.The catalyst provided a FEco of 94% at −0.58 V (vs. RHE). It was found that the vacancies in graphite oxide affected the electronic structure of the anchored Fe-N4sites. The edgeanchored Fe-N4sites exhibited a downward shift of the d-band center compared to those Fe-N4sites immobilized on an intact graphene support, which weakened the binding strength of *CO on the active site. The defects shortened the Fe-C bond length formed between the active site and *CO, thereby improving the ECR performance.
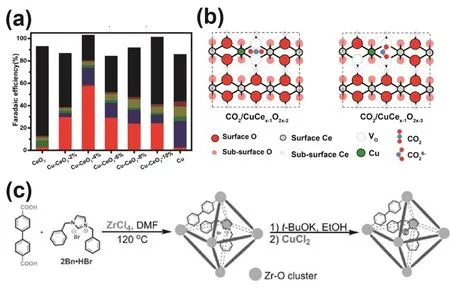
Fig. 8 (a) FEs of different products for Cu-CeO2 with different Cu concentrations. (b) Structure models of the Vo-bound and single-atom Cu sites on CeO2 for CO2 adsorption and activation.(c) Schematic of the synthesis of 2Bn―Cu@UiO-67.
5.3.2 Non-coordination heteroatom doping
The strategy of non-coordination doping is mainly used in carbon-based SACs with nonmetal elements such as B, P, S, Cl,and F, where the electronegativity difference can modulate the electronic structures of the active sites, thereby improving the electrocatalytic activity158–161. These heteroatoms can perturb the electron arrangement around the central element, and change the physical properties of the hosts such as the porosity and electrical conductivity. Sulfur is commonly used for doping into carbon substrates, which owing to its large atomic radius tends to be attached at carbon defects. For instance, Li and co-workers utilized FeCl3as the metal precursor and added ethylenedioxythiophene (EDTO) and acetonitrile followed by pyrolysis to obtain an Fe, N, S coexisting carbon layers (Fe-NS-C) with Fe-N4active sites160. The as-made catalyst can be used for ECR to achieve a FEco of 98% at an overpotential of 0.47 V. Addition of EDTO was observed to induce more defects as well as larger specific surface areas. XPS and XANES characterization showed that Fe atoms in the Fe-NS-active centers existed in an oxidized state. By performing DFT calculations, S doping was proposed to raise the Fe 3d orbital toward the Fermi energy, enriching the electron density of the Fe center and further decreasing the energy barrier for the generation of *COOH.
In addition to S atoms, F has also proved to be an effective non-coordinating doping element to regulate the electronic structure of Ni-N4centers. For example, Zhu and co-workers synthesized a Ni-N4catalyst with F doping by employing a polytetrafluoroethylene (PTFE)-assisted pyrolysis method158.The addition of PTFE favored the formation of ultrathin nanosheet morphologies. In the synthesized materials, the incorporated F atoms raised the energy of the Ni 3d orbital electrons closer to the Fermi level, which decreased the formation energy of *COOH on its surface. The catalyst achieved a FEco of 95% at applied voltages ranging from −0.67 to −0.97 V (vs. RHE). The focus of this non-coordination heteroatom doping is to modify the electron density of the M-Nxactive sites, potentially decreasing the energy barrier for the generation of CO2reduction intermediates.
5.3.3 Spatial structure modulation
Increasing the density of metal active sites on the catalyst surface provides an effective means to enhance the catalytic activity. To this end, achieving high metal loadings is useful,however it is challenging because of the easy aggregation of metals at high loading contents. Keeping the metal loading constant, increasing the specific surface areas of the support provides more sites for anchoring single atoms. For instance, Li and co-workers employed N-doped carbon substrates of different sizes for constructing Ni SACs162. The 120 nm-sized morphology was found to facilitate the formation of more active metal sites due to its abundant exposed micropores and high nitrogen contents. The as-obtained Ni SACs exhibited a CO partial current density of 726 mA·cm−2in a flow cell. In another work, Chen and co-workers prepared an Fe SAC with Fe-Nxsites immobilized on mesoporous carbon nanoframes by pyrolysis of Fe-doped MOF precursors163. During the pyrolysis process, the isolated Fe2+in the ZIF precursors induced a Kirkendall effect, favoring the formation of mesoporous carbon nanoframes with hierarchical pore sizes, thus improving the availability of single Fe atoms for active ECR. This sacrificial template method is widely utilized to synthesize carbon substrates with high specific surface area.
In addition, the spatial confinement strategy can facilitate the multi-electron reduction of CO2by creating tiny reaction spaces.Chen et al. reported a N-heterocyclic carbene (NHC)-ligated copper single atom site (Cu SAS) embedded in a metal-organic framework (2Bn-Cu@UiO-67)164(Fig. 8c). This Cu SAS gave a CH4FE of 81% via the ECR at −1.5 V (vs. RHE) with a current density of 420 mA·cm−2. It was proposed that the electrondonating effect of nitrogen heterocyclic carbene ligands enhanced the charge density of Cu sites, thus optimized the adsorption of *CHO. Meanwhile, the pore structure of UiO-67 possessed a strong CO2capture ability, and the narrow space between the pores promoted the diffusion of the reduction intermediates between the active sites. These provided favorable aspects for generation of CH4.
5.4 Surface functionalization
For heterogenized molecular SACs, side chains or functional groups can be easily introduced to control their catalytic performance165. Stimulated by the natural oxygen-evolving complex (OEC) of photosystem II, electronwithdrawing/donating species can be introduced onto the side chain of molecular SACs to boost ECR activity166. The introduced electron-withdrawing/donating groups are supposed to regulate the redox-mediated ECR mechanism by modulating the redox potential of the active sites. For instance, Huang and co-workers modified CoPc/G with an electron-donating amino group (amino-CoPc/G) and electron-withdrawing nitro group(nitro-CoPc/G), and further studied their effects on ECR167.Electrochemical tests illustrated that nitro-CoPc/G provided higher ECR performance to yield CO. By DFT calculations, the generation of *CO2−was inferred to be the RDS. After introduction of the electron-withdrawing nitro group, the electron density near the Co atoms was lowered. Consequently,the dz2 orbital upshifted, and became less influenced by the other low energy orbitals (dxy). The energy difference between dz2 and dxyis negatively correlated with the energy barrier to form *CO2−.Similar to the nitro group, the introduction of electronwithdrawing cyano (CN) groups into CoPc was also observed to enhance the ECR ability to produce CO168. Besides electronwithdrawing moieties, an electron-donating methoxy species was introduced into the Pc ligands to enhance the catalytic performance of NiPc169. This improvement was attributed to the partial reduction of the Ni centers with the addition of electrondonating moieties.
6 Conclusion and perspectives
SACs are showing an enormous promise for the realization of high-efficiency ECR with the potential to attain 100% dispersion and utilization. A range of supported SACs have been demonstrated to catalyze the ECR especially to C1hydrocarbons(oxygenates), with superior activity and selectivity compared to traditional heterogeneous electrocatalysts. SACs with d-block elements such as Fe, Ni, Mn, Mo, Ru, Rh, Pd, Ir, and Pt can convert CO2to CO. SACs with ds-block metals such as Cu and Zn enable deep reduction of the ECR to produce CH3OH and CH4, respectively. Formate is the major reduction product for pblock SACs. Among these metals, transition metals, especially Fe, Co, Ni, Cu, and Zn, are the most frequently used because of their low cost and earth abundance. Another class of SACs are conjugated macrocycles (CM) such as porphyrin and phthalocyanine-based complexes. They have well-defined structures, thus enabling the construction of accurate structural models for the study of the ECR mechanism and understanding of the structure-performance relationship. Nevertheless, singleatom catalysis of CO2reduction is still in its early stage. To move this forward, in addition to screening and identification of novel atoms with more-advantageous properties for ECR, further progress in controlled synthesis and characterization is desirable.In this regard, more work is required on developing schemes to enable ultra-high dispersion and loading of SACs with precise control in their composition and coordination structure as well as their interaction with the host170. Equally importantly,scalable synthesis methods with low cost and mild operation conditions are required to achieve commercialization. To discover and design new and advanced SACs for ECR,application of computational screening coupled with machine learning would be helpful148.
Single atoms are inclined to aggregate into clusters or nanoparticles via Ostwald ripening. In addition, under ECR reaction conditions, SACs may undergo coordination structure alteration particularly at negative applied voltages, such as the reversible conversion between single atoms and nanoparticles,adsorption of reaction intermediates, and leaching in to the electrolytes. However, in many prior references, the structureactivity relationship is established by characterization and theoretical simulations under ideal conditions. The real active centers during the catalytic process are still ambiguous.Therefore, it is necessary to probe and monitor the dynamic behaviors and structure evolution of SACs during the ECR by using operando/in situ characterization techniques such as liquid-phase electron microscopy, scanning tunneling microscopy, XAS and EXAFS. Besides, operando infrared spectroscopy and Raman spectroscopy, online electrochemical mass spectrometry, and operando electron paramagnetic resonance are invaluable tools for tracking the evolution of reaction intermediates. They also provide unique fingerprints of bound molecules, indicative of the type of surface sites. These will help create more accurate theoretical models and gaining a deeper insight into understanding of the ECR reaction pathways,degradation mechanisms as well as catalytic structure-property relationships specific to a given SAC, further guiding new direction for the design of SACs. Developing methods to stabilize SACs against sintering and structure change especially under industrially relevant ECR conditions should be the focus of interest for future study.
Note that the ECR products by single-atom catalysis are limited, solely including CO, HCOOH, CH4and only a small amount of C2+products. Considering C2+products and liquid hydrocarbons are more valuable for industrial use, developing SACs that are active and selective for multicarbon product formation is meaningful but very challenging. To date, Cu-based SACs are the most competitive candidates for C-C coupling reactions. It is believed that construction of diatomic sites,especially incorporation of a second metal single atom(s) on the surface of Cu-based hosts, is likely an effective strategy to afford cascade catalysis to facilitate the deep reduction of CO2by tuning ECR intermediate adsorption energy as well as hydrogen binding strength. Alternatively, design of a combined system consisting of SACs and an ensemble of trimers or a few atoms or clusters or nanoparticles permits increased metal loadings and also provides adjacent atoms of defined chemical identity to increase the catalyst versatility. This is expected to greatly enhance reduction current density107.
