绵羊NDRG1 基因表达和干扰载体的构建及验证
2022-11-17龙德智孙楠祯郝科兴陈慧慧胡广东
龙德智,孙楠祯,郝科兴,陈慧慧,王 静,胡广东
(石河子大学动物科技学院,新疆石河子 832000)
N-myc 下游调节基因1(N-myc Downstream Regulated Gene 1,NDRG1)是一种蛋白大小为43 Kda,广泛分布于哺乳动物组织中的转移抑制因子。研究表明,该基因可参与包括NF-κB 通路[1]、TGF-β通路[2]和Wnt/β-catenin等信号通路的调控过程[3],在多种转移相关的生物学功能中发挥重要作用[1]。研究表明,NDRG1通过Wnt/β-catenin通路调节钙粘蛋白E(E-Cadherin)和基质金属蛋白酶(MMPs)使细胞发生迁移或侵袭[4-5]。因此,NDRG1 被认为是一种信号通路的中央调节剂。此外,NDRG1基因在妊娠小鼠胚胎附植过程中表达量显著升高,深入研究发现,NDRG1表达变化会影响胚胎的粘附及侵袭过程,进而调控胚胎附植成败[6]。
目前,关于NDRG1基因影响胚胎附植机制研究主要集中在人和小鼠上,与绵羊胚胎附植相关研究尚未见报道,而研究该基因对绵羊胚胎附植机制的调控,需要构建可靠的转基因载体。因此,本文以绵羊NDRG1基因为研究对象,利用RT-PCR 扩增绵羊NDRG1基因CDS 区,构建表达和干扰载体并确认其在细胞中的表达情况,为研究NDRG1基因在绵羊细胞中的作用提供研究材料。
1 材料与方法
1.1 实验材料 HEK 293T 细胞、pcDNA3.1(+)和pGBplv-U6-mcherry-puro 质粒由本实验室保存,pGM-T 载体购自Promega 公司(美国),绵羊滋养层细胞由本实验室利用组织块分离法获得。
1.2 主要试剂 Trizol 购自Invitrogen 公司(美国),T4 DNA 连接酶(TaKaRa,中国大连)、PrimeScriptTMRT-PCR Kit(TaKaRa,中国大连)购自宝生物工程(大连),HIFI DNA Taq 聚合酶购自全式金生物公司(中国北京),OMEGA 质粒提取试剂盒(美国)购自上海生工生物工程有限公司,凝胶回收试剂盒购自Axygen生物技术有限公司(美国),大肠杆菌DH5α购自TIANGEN 公司(中国北京),DMEM 购自GIBCO 公司(美国),BamHI 和EcoRI 购自NEB 公司(美国),脂质体转染试剂(Lipofectamine 2000 ReAgent)购自Invitrogen 公司(美国),兔抗NDRG1 和鼠抗β-actin抗体购自碧云天公司(中国上海)。
1.3 引物设计与合成 根据NCBI 已公布预测的绵羊NDRG1基因序列(登录号:XM_027972876.2),利用Primer 5 软件设计NDRG1 CDS 区扩增引物和qPCR 检测引物,扩增引物上下游分别引入BamHI 和EcoRI 酶切位点(表1);根据在线靶点设计软件设计NDRG1-RNAi 片段,RNAi 片段添加Loop 环序列和KpnI 酶切位点并在上下游分别添加AgeI 和EcoRI 酶切位点(表1),送上海生工生物工程技术公司合成。

表1 实验所用引物及序列
1.4NDRG1基因CDS 区克隆 以绵羊滋养层细胞为材料,利用Trizol 法提取总RNA,利用反转录试剂盒进行反转录,合成cDNA 链。以制备的cDNA 为模板,PCR 扩增NDRG1基因CDS 区全序列。PCR 反应体系(50 μL):10×Buffer 5 μL,LA Taq 酶0.5 μL,dNTP 4 μL,上下游引物各1μL(10 μmol/L),cDNA 模板2 μL(1 μg/μL),补水至50 μL。PCR 反应程序:98℃预变性2 min;98℃变性10 s,退火10 s,72℃延伸10 s,30 个循环;72 ℃延伸10 min,4 ℃保存。扩增产物用1%的琼脂糖凝胶电泳检测,回收目的片段与pGEM-T Vector 载体连接,将连接产物转化至DH5α感受态细胞中,过夜培养挑取单菌落,扩大培养后提取质粒,命名为pNDRG1-T,由上海生工生物工程技术公司进行双向测序。利用DNAMAN 软件将测序结果与NCBI 已公布预测的绵羊NDRG1基因序列进行序列比对。
1.5 NDRG1 表达载体构建 以重组质粒pNDRG1-T 和pcDNA3.1(+)为模板,利用BamHI 和EcoRI 进行双酶切,分别胶回收双酶切后的NDRG1基因目的片段和pcDNA3.1(+)骨架载体,利用T4 DNA 连接酶于4℃水浴过夜连接,转化至DH5α感受态细胞中,挑取单菌落,扩大培养,提取质粒,双酶切鉴定,构建表达载体命名为pcDNA3.1-NDRG1。
1.6 NDRG1 干扰载体构建 将合成的干扰片段稀释至50 μmol/L,取4.5 μL 的正义链和反义链寡核苷酸(oligo)序列和1 μL 退火buffer,于93℃ 3 min 进行退火反应,降至室温,-20℃保存待用。以pGBplv-U6-mcherry-puro 质粒为模板,应用限制性内切酶AgeI 和EcoRI 剪切,胶回收双酶切后的载体骨架。将酶切后线性化的pGBplv-U6-mcherry-puro 与退火后的oligo 片段连接,反应体系如下:ligation Mix 7.5 μL,回收的干扰载体1 μL,退火oligo 片段6.5 μL,总体系为15 μL,于恒温水浴系统4 ℃过夜连接。将连接产物转化至DH5α感受态细胞中,过夜培养挑取单菌落,扩大培养后提取质粒,分别命名为NDRG1-shRNA1、NDRG1-shRNA2、NDRG1-shRNA3 和NDRG1-shRNA4。
1.7 重组质粒酶切鉴定 将抽提好的表达重组载体(pcDNA3.1-NDRG1)利用BamHI 和EcoRI 进行双酶切鉴定,干扰重组载体(NDRG1-shRNA1、NDRG1-shRNA2、NDRG1-shRNA3 和NDRG1-shRNA4)利用KpnI 进行单酶切鉴定,于37℃恒温水浴系统反应1 h后凝胶电泳鉴定。鉴定结果无误进行无内毒素质粒抽提以获得足够量重组质粒,-20℃保存。
1.8 重组载体基本功能验证
1.8.1NDRG1基因表达载体基本功能验证 快速溶解HEK293T 于24 孔板加入含10%FBS 和1% 双抗的DMEM 完全培养基,在37℃、5%CO2细胞培养箱中培养,待细胞密度达70%~80%可进行转染。设置空白组、C1 对照组(pEGFP-C1)和过表达组(pcDNA3.1-NDRG1),利用Lipofectamine 2000 ReAgent 脂质体进行质粒转染(相关过程参见说明书)。转染48 h 后检测NDRG1 转录水平,利用Trizol 法提取总RNA,使用Prime ScriptTMRT-PCR Kit 将样品RNA 反转录成cDNA,qRT-PCR 分析备用。qRT-PCR 反应体系:10 μL SYBR Premix Ex Taq TM II(2×),PCR上下游引物(10 μmol/L)各0.8 μL,2 μL cDNA 模板(50 ng/μL),6.4 μL ddH2O。qRT-PCR反应条件:95℃50s;95℃5s,60℃30s,40个循环;95℃15s,60℃1min,95℃ 15 s。以β-actin基因为内参基因,采用2-ΔΔCt法计算NDRG1基因的相对表达量(PCR 引物序列表见表1),各组实验至少重复3 次。转染48 h 后检测NDRG1 蛋白表达水平,收集细胞利用细胞裂解液(由RIPA 裂解缓冲液和PMSF 蛋白酶抑制剂组成)重悬细胞,冰上裂解5 min。加入等量4×SDS-蛋白上样缓冲液,置于100℃水浴锅中煮沸10 min。将提取的细胞蛋白进行SDS-PAge凝胶电泳,各组蛋白上样量均为20 μL,选用Blue Plus II Protein Marker。经过转膜、封闭、一抗孵育、二抗孵育,在暗室中使用Western Bright ECL 化学发光底物进行显色反应并用胶片曝光,用ImAgeJ 软件对目的条带进行灰度分析。
1.8.2NDRG1基因干扰载体基本功能验证 转染设置表达组(pcDNA3.1-NDRG1)、干扰载体对照PLV 组(pcDNA3.1-NDRG1 和pGBplv-U6-mcherry-puro)、N1 组(pcDNA3.1-NDRG1 和NDRG1-shRNA1)、N2组(pcDNA3.1-NDRG1 和NDRG1-shRNA2)、N3 组(pcDNA3.1-NDRG1 和NDRG1-shRNA3)和N4 组(pc DNA3.1-NDRG1 和NDRG1-shRNA4)。转 染48 h 后检测NDRG1 转录水平和蛋白的表达,方法见1.8.1。
1.9 统计分析 应用SPSS 统计软件进行数据分析。采用单因素方差分析比较各处理组间NDRG1 转录水平及蛋白表达水平的差异,以LSD-t 检验法进行组间两两比较。“*”为P<0.05,“**”为P<0.01,“***”为P<0.001,P<0.05 即为显著。
2 结果与分析
2.1NDRG1基因CDS 区的获取与序列分析 扩增产物经1% 琼脂糖凝胶电泳鉴定,大小为1 378 bp(图1A),与NCBI 比对结果一致,TA 克隆载体双酶切鉴定结果显示目的片段大小与预期一致(图1B)。绵羊NDRG1基因CDS 区核苷酸编码氨基酸384 个,序列进行比对结果显示,绵羊扩增片段与预测片段相比1 031位点存在一个G →A 碱基突变(图1C),但氨基酸比对完全相同(图1D),以上说明绵羊NDRG1基因CDS 区克隆成功。
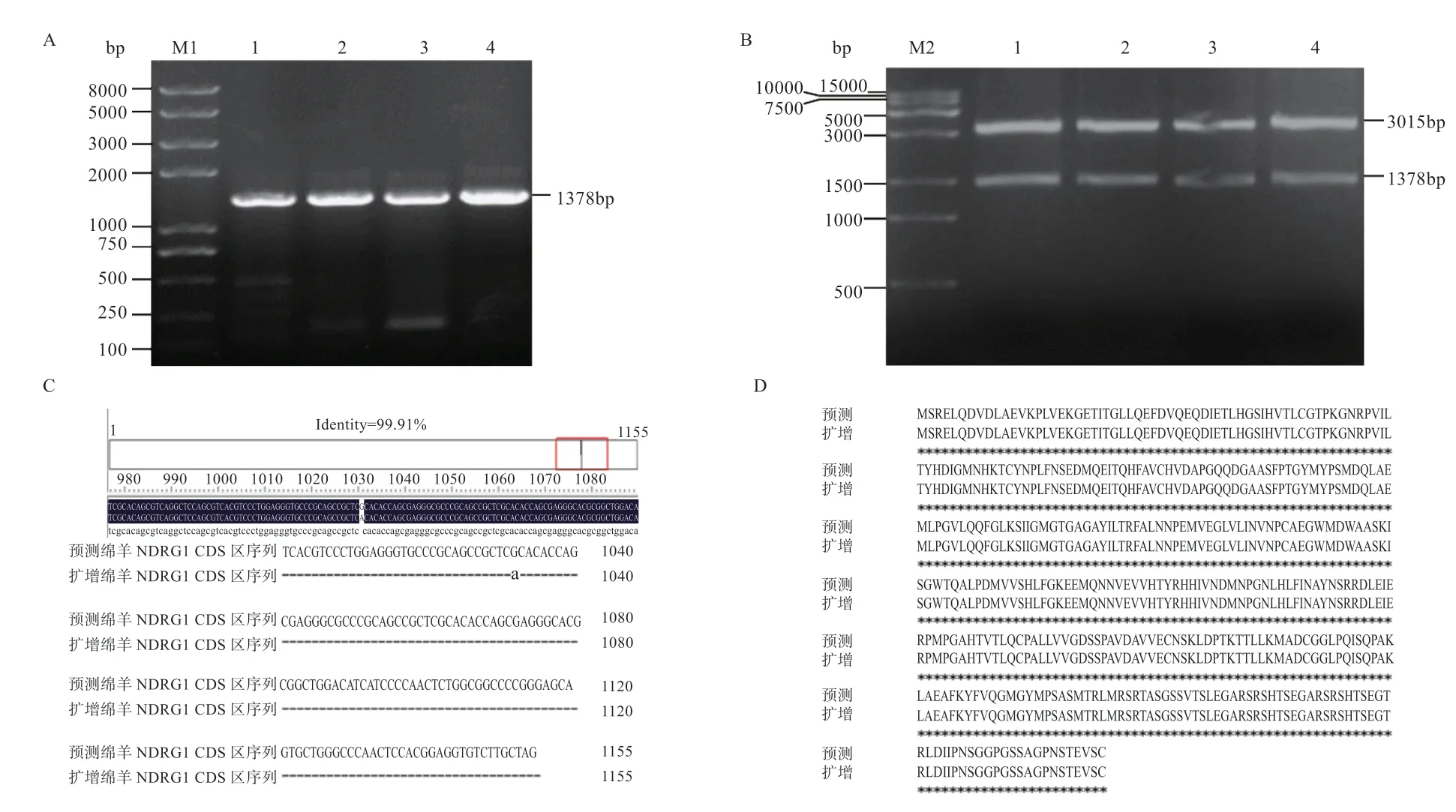
图1 NDRG1 基因克隆及鉴定
2.2 重组表达和干扰载体酶切鉴定结果 用BamHI 和EcoRI 双酶切重组表达载体pcDNA3.1-NDRG1,得到2 条大小约为5 428 bp 和1 378 bp 的条带(图2A),结果与预期相符,证明重组表达载体构建成功。用KpnI分别对4 条重组干扰载体NDRG1-shRNA1、NDRG1-shRNA2、NDRG1-shRNA3 和NDRG1-shRNA 4进行单酶切,得到2 条大小约5 651 bp 和2 041 bp 的两条带(图2B),结果与预期相符,证明4 条重组干扰载体构建成功。

图2 重组质粒酶切鉴定
2.3 重组表达质粒在HEK293T 细胞中的功能验证 采用脂质体转染的方法将重组质粒pcDNA3.1-NDRG1 和pEGFP-C1 导入HEK293T 细胞中,转染24 h 后荧光观察结果见图3,结果初步判断重组质粒导入HEK293T细胞中。qRT-PCR结果显示,pcDNA3.1-NDRG1 组NDRG1基因的表达水平极显著高于空白对照组和C1对照组(图4A);Western Blot 结果显示,出现了2条清晰的大小分别为43 kDa 和42 kDa 的蛋白条带,结果与预期相符,pcDNA3.1-NDRG1 组NDRG1 蛋白表达水平极显著升高(图4B),上述结果表明,重组表达质粒pcDNA3.1-NDRG1 能正常在细胞中表达。
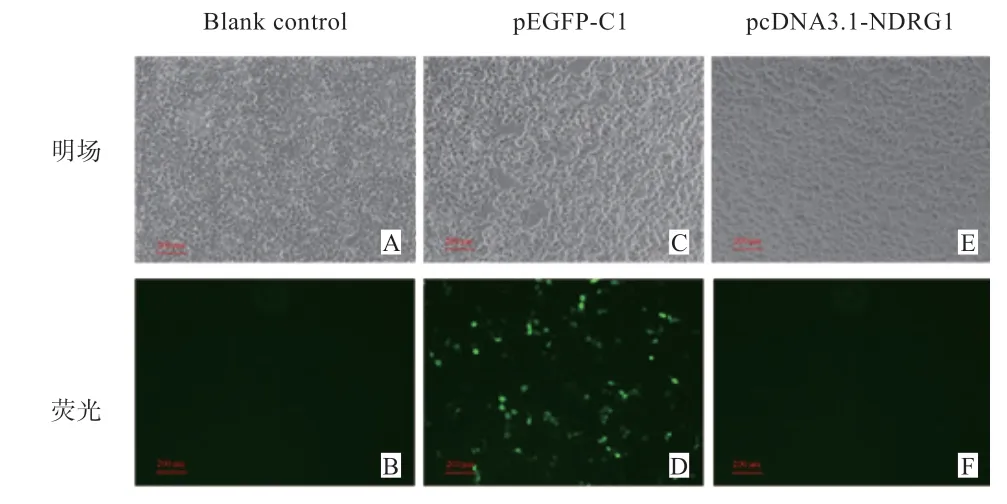
图3 重组表达载体荧光鉴定(200 μm)

图4 重组表达载体mRNA 和蛋白相对表达水平
2.4 有效干扰片段筛选结果 脂质体转染24 h 后观察荧光图为图5,结果显示重组干扰质粒成功转入HEK293T 细胞中。qRT-PCR 结果显示,PLV 组与pcDNA3.1-NDRG1组相比NDRG1基因表达无显著性差异;干扰组N1、N2、N3 和N4 组与pcDNA3.1-NDRG1 组相比NDRG1基因表达量均显著或极显著降低,N3 组NDRG1基因表达量最低,N1、N2、N3 和N4 组NDRG1基因相对表达量分别为36%、61%、30% 和79%,其干扰效率分别为64%、39%、70% 和21%。Western Blot 结果显示,PLV 组与pcDNA3.1-NDRG1 组相比蛋白表达水平无显著性差异,N1、N2、N3 和N4 组与pcDNA3.1-NDRG1 组相比蛋白表达水平均极显著降低(图6),N1、N2、N3 和N4 组NDRG1 蛋白相对表达量分别为9.4%、11%、9.2%和61%,其干扰效率分别为90.6%、89%、90.8%和39%,根据NDRG1 的表达水平筛选出N3 组干扰效果最好,即NDRG1-shRNA3 为最佳干扰载体(图7)。

图5 重组干扰载体荧光鉴定(400 μm)

图6 重组干扰载体mRNA 和蛋白相对表达水平

图7 重组干扰载体干扰效率
3 讨 论
NDRG 家族中有4 个成员,即NDRG1、NDRG2、NDRG3和NDRG4,NDRG1是其中最早发现的基因[7],NDRG 家族蛋白结构相似度极高,有共同的α/β水解酶折叠结构域,但NDRG1 更为特殊,其C 端(339-369残基)存在10 个氨基酸组成的3 个串联重复的序列(GTRSRSHTSE)[8],这使得其能够与金属离子结合并参与应激反应和细胞保护的生理功能相关[9-12]。NDRG1是一种能够普遍表达于多种组织中的基因,但是其表达方式更具有选择性,NDRG1 mRNA 能够在心脏、卵巢、骨骼肌、结缔组织和血管中表达,但其蛋白却未能在这些组织中检测出来,更深入的研究发现NDRG1 的亚细胞定位因组织的类型而不同,而细胞定位决定了其在细胞的功能,这也说明NDRG1 在不同细胞会行使不同的功能[13-14]。
关于绵羊NDRG1 表达载体和干扰载体的构建在国内未见报道,本实验利用绵羊滋养层细胞cDNA,通过RT-PCR 的方法成功扩增出NDRG1基因CDS 区序列。经比对显示克隆的绵羊NDRG1 编码区在1 031 位点存在一个由G 突变为A 的单碱基突变,但氨基酸比对发现二者氨基酸完全相同,并未引起氨基酸序列的改变。核苷酸的差异可能来自于核苷酸的多态性,但并不影响蛋白的表达,克隆的绵羊NDRG1基因可用于后续实验。
实验中将克隆的NDRG1 序列插入pcDNA3.1+载体构建表达载体,设置空白对照组,同时由于pcDNA3.1+不含荧光标记基因故设置了C1 对照检验细胞的转染效率。RNAi 技术是用双链RNA(Double-Stranded RNA)引起真核生物体内同源序列mRNA 特异性降解从而在转录和翻译水平上阻断相关基因表达使基因沉默,根据其来源分为内源性siRNA(endo-siRNA)和外源性siRNA(exo-RNA)[15]。由于siRNA 在细胞中易分解并不能长期存在,故实验选择构建shRNA 干扰载体的方法达到干扰基因的目的。实验中构建了4 个干扰载体分别为NDRG1-shRNA1、NDRG1-shRNA2、NDRG1-shRNA3 和NDRG1-shRNA4,干扰的靶点位置分别为113、214、426 和602。干扰载体转染结果显示重组干扰载体成功导入293T 细胞,实验结果显示4 个干扰载体都可以抑制NDRG1 蛋白在HEK293T 细胞的表达,即设计的干扰片段能靶向结合NDRG1基因113、214、426 和602 位点从而达到抑制基因表达的作用,其中靶点位置426 的干扰载体(NDRG1-shRNA3)干扰效果最好,干扰效率的差异和位点的不同有关[16]。
NDRG1 因为抑制肿瘤生长和转移的能力受到关注,继被确定为结肠癌转移因子后又证明NDRG1 能在其他组织中起到抗肿瘤和抗转移能力[13]。Kachhap 等[17]深入探究发现其原理是NDRG1 能够参与囊泡在核周区域和细胞表面之间的循环,即含有NDRG1 的囊泡与含有E-钙粘蛋白(E-cadherin)的囊泡融合,使E-cadherin循环到细胞膜上影响上皮间质转化继而影响癌细胞的侵袭与迁移。哺乳动物胚胎的着床方式为表面着床,胚胎滋养层仅与子宫腔上皮细胞接触并在接触位点发生粘附,细胞增殖、迁移,血管发生等生物学行为[18]。妊娠的发生从一定角度上来说与癌细胞的转移大相径庭,其过程都发生细胞的转移。Lachat 等[14]通过对大量组织进行免疫荧光和免疫组化证明NDRG1 mRNA 和蛋白能够在胎盘组织中表达,NDRG1 在子痫孕妇胎盘中能够差异性表达,并通过细胞外调节激酶(ERK)/基质金属蛋白9(MMP-9)通路影响滋养层细胞的侵袭,同时NDRG1 的上调通过PI3K/AKT 抑制人胎盘的血管生成,研究人员也证明NDRG1 表达的降低会导致小鼠妊娠失败[6,19-20]。为探究NDRG1基因在绵羊妊娠过程中的具体作用,本实验构建NDRG1 的表达及干扰载体为后续实验提供实验材料。
4 结 论
本研究成功构建了绵羊NDRG1基因表达载体和干扰载体,为进一步研究NDRG1基因在绵羊中的调控机制提供材料。
