白花刺续断野生居群的叶绿体全基因组特征解析
2022-11-16张德全
张 倩, 张德全,2*
( 1.大理大学 药学院, 云南 大理 671000; 2. 云南省滇西抗病原植物资源筛选研究重点实验室, 云南 大理671000 )
白花刺续断(Acanthocalyxalba),又名白花刺参,隶属于川续断科(Dipsacaceae)刺续断属(Acanthocalyx)。该属在我国有4种2变种,主要分布在云南、四川、西藏等地(Hong et al., 2011)。白花刺续断以全草入药,为传统藏药,其藏药名为“江才嘎保”,始载于《四部医典》(国家中医药管理局中华本草编委会,2002)。它是国家卫生部藏药药品标准中收载的三种“刺参”之一(青海省药品检验所和青海省藏医药研究所,1996),具有健胃、催吐之功效。内服可用于关节疼痛、小便失禁、腰痛、眩晕及口眼歪斜,外用治疗疮、化脓性创伤,还具有抗肿瘤作用(国家中医药管理局中华本草编委会,2002;杨竞生,1989)。近年来,有关白花刺续断的研究主要集中在其有效成分、含量测定及提取工艺等方面(吴春蕾等,2011;Zhang et al., 2013;杨圣贤等,2014;张志锋等,2015)。如张志锋等(2018)发现白花刺续断中含有皂苷、生物碱、甾醇等类型化合物,其中皂苷类是其主要有效成分。而分子生物学方面的研究较少,仅有Wang等(2020)报道了该种的叶绿体基因组序列。那么,在白花刺续断的种内居群水平上,其叶绿体基因组序列有什么变化特征呢?
叶绿体基因组在被子植物中通常为母系遗传。与核基因组和线粒体基因组相比,其基因进化速率慢,且在基因组成及结构方面比较保守(Smith, 2015;Szymon et al., 2016;Du et al., 2020),这使得叶绿体基因组在植物物种鉴定和谱系进化研究中具有重要作用。Cui等(2019)对比分析了32种豆蔻属(Amomum)植物叶绿体基因组,结果表明叶绿体全基因组可准确鉴定豆蔻属物种;李依容等(2020)利用叶绿体基因组揭示了民族药滇白珠(Gaultherialeucocarpavar.yunnanensis)复合群的系统发育关系;Zhang等(2021)基于叶绿体基因组重建了桃金娘目(Myrtiflorae)的物种分化时间和系统发育关系。但叶绿体基因组在长期进化过程中,结构上会发生一些变异,如反向重复区收缩、倒位、基因和内含子的丢失等(Zhang et al., 2014;Liao et al., 2020;姜汶君等,2020),这些结构变异为揭示物种系统发育和进化关系等提供了遗传信息。由此可见,植物叶绿体全基因组序列能提供丰富的遗传信息,在分类鉴定、系统发育及进化等方面具有重要意义。那么,植物叶绿体全基因组是否适用于种内居群水平上的群体遗传学研究?由于群体水平上测序成本较高,数据分析方法尚不成熟等,相关研究还较少。
本研究以白花刺续断野生居群个体为研究材料,拟采用二代测序技术进行高通量测序,并对其叶绿体全基因组进行拼接、注释及进化分析。拟探讨以下科学问题:(1)白花刺续断的叶绿体全基因组序列有何特征;(2)叶绿体全基因组能否用于解析白花刺续断种内居群水平上的遗传结构。本研究将为刺续断属相关物种的分子遗传学研究奠定基础,也为叶绿体全基因组在群体遗传研究方面开展初步尝试。
1 材料与方法
1.1 实验材料
本实验中,白花刺续断分子材料采自于四川省甘孜州的5个野生居群(表1),共10份实验材料。经大理大学张德全教授鉴定为白花刺续断(Acanthocalyxalba),其凭证标本保存于大理大学药学院药用植物与生药标本馆。

表 1 白花刺续断样品采集信息Table 1 Collection information of Acanthocalyx alba samples
1.2 基因组DNA提取与测序
DNA提取采用改良的CTAB方法,从硅胶干燥的叶片材料中提取总基因组DNA。利用Covaris超声波破碎仪将基因组DNA片段化,经末端修复、加A尾、加测序接头、纯化及PCR扩增等过程,构建测序文库。文库经质量检测合格后,使用带有双末端(pair-end) (2×300 bp) Illumina Hiseq 2 500平台进行二代测序,整个测序工作在北京诺禾致源生物科技有限公司完成。
1.3 叶绿体全基因组的组装、注释
经二代测序,得到4 G左右原始数据(Raw Data),经Trimmomatic V.0.32过滤处理后,利用GetOrganelle.py进行组装,后续数据处理参考本课题组前期工作(胡海粟和张德全,2021)。以白花刺续断(序列号:NC_045055)为参考基因组,使用Geneious 8.0.2软件完成白花刺续断叶绿体全基因组的注释,并将其提交到NCBI数据库(https://www.ncbi.nlm.nih.gov/),利用在线工具Organellar Genome Draw(https://chlorobox.mpimp-golm.mpg.de/OGDraw.html)绘制叶绿体基因组物理图谱。
1.4 IR/SC边界收缩扩张及SSR分析
将注释好的10条基因组序列上传至网站IRscope(https://irscope.shinyapps.io/irapp/)进行IR边界的收缩和扩张分析,最后得到的图片采用绘图工具Adobe Illustrator CC 2015进行人工调整。同时,利用MISA软件搜索简单重复序列(simple sequence repeats,SSR)位点并分析白花刺续断叶绿体基因组的SSR特征。
1.5 序列差异比较分析
在采用MAFFT V.7.129软件对10条白花刺续断叶绿体基因组序列进行比对后,使用BioEdit软件手动调整序列。使用DnaSP V.7.0.26对叶绿体基因组中的核苷酸变异性(Pi)进行滑动窗口分析。步长设置为200 bp,窗口长度为600 bp。P-distance使用MEGA v.7.0.26软件进行统计分析。此外,将叶绿体基因组序列的注释进行格式转换,利用在线软件mVISTA(http://genome.lbl.gov/vista/mvista/submit.shtml)的Shuffle-LAGAN模式对白花刺续断叶绿体全基因组比较分析,选取Acanthocalyxalba(NC_045055)作为参考序列。
1.6 系统发育分析
根据白花刺续断叶绿体基因组注释信息,从NCBI数据库下载已发表的川续断科及忍冬科(Caprifoliaceae)的11种植物的叶绿体全基因组序列,用于系统发育分析。选择小粒咖啡(Coffeaarabica)和中粒咖啡(C.arabica)为外类群,使用MAFFT V.7.1将白花刺续断与下载的叶绿体全基因组序列进行多序列比对。系统发育树的构建,采用了最大似然法(maximum likelihood, ML)、最大简约法(maximum parsimony, MP)和贝叶斯推论法(Bayesian inference, BI)3种方法。核苷酸替代模型经jModelTest V 2.1.7软件筛选定为GTR+G模型。利用RAxML V.8.2.4软件构建ML系统树,采用快速靴带算法,重复1 000次。利用MEGA V.7.0.26软件构建MP树,重复1 000次。利用MrBayes V.3.2.6构建BI树,基于马尔科夫链蒙特卡洛(MCMC)算法,计算100万代,每隔1 000代取样一次,舍弃前25%棵树,根据剩余的样本构建一致树。
2 结果与分析
2.1 基因组结构与基本特征
白花刺续断的叶绿体全基因组为常见的四分体结构,由两个反向重复区IRs(inverted repeats)、一个大单拷贝区LSC(large single copy)和一个小单拷贝区SSC(small single copy)组成(图1,表2)。拼接后的白花刺续断叶绿体基因组,全长为155 335~156 266 bp,GC含量为38.1%~38.2%。各区段长度分别为89 027~89 076 bp(LSC)、17 689~17 842 bp(SSC)、24 253~24 666 bp(IRs)。4个区段中GC含量最高的是IR区(42.8%~43.2%),其次是LSC区(36.5%)和SSC区(32.9%)。经注释,得到113个基因,包括72个编码蛋白基因、30个tRNA基因、4个rRNA基因和7个假基因(clpP、accD、ycf2、ycf1、rps18、rps3和ycf3)。此外,白花刺续断叶绿体全基因组中有16个基因含有内含子(intron),且均只含有一个内含子(表3)。
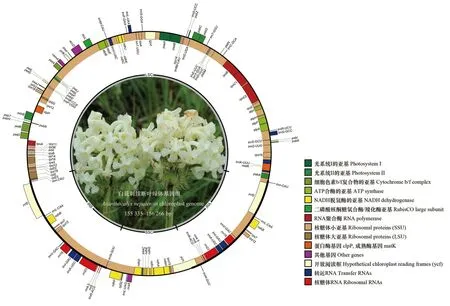
圆圈外的基因是顺时针转录,圆圈内的基因是逆时针。图中颜色表示功能基因。内部的深灰色对应于GC含量,浅灰色对应于AT含量。The genes outside the circle are transcribed clockwise, while the genes inside the circle are transcribed counterclockwise. The colors here represent functional genes. The inner dark gray corresponds to the GC content and the light gray corresponds to the AT content.图 1 白花刺续断叶绿体全基因组图谱Fig. 1 Gene map of complete chloroplast genome in Acanthocalyx alba

表 3 白花刺续断叶绿体全基因组基因组成Table 3 Composition of complete chloroplast genome of Acanthocalyx alba
2.2 IR/SC边界收缩扩张及SSR分析
叶绿体基因组由两个反向重复的IR区、LSC区与SSC区构成,因此存在LSC/IRb、IRb/SSC、SSC/IRa和IRa/LSC 4个边界。在基因组进化过程中,4个边界会发生扩张与收缩,使某些基因进入IR区或单拷贝区。不同地点白花刺续断的叶绿体基因组的4个边界相对保守(图2)。LSC/IRb边界在白花刺续断中位于rpl23基因内部,且位于LSC区域的差异不大,为185~186 bp;SD01、YD01、YD02、LT01、LT02、KD01、KD02的IRb/SSC边界基因完全相同,位于IRb区trnN-GUU基因138 bp处,而SD02、DF01、DF02的trnN-GUU基因扩张到SSC内部,距离IRb/SSC边界48~223 bp;SSC/IRa的边界在白花刺续断基因组中都位于ycf1基因内部;IRa/LSC边界全部位于trnH-GUG基因附近。
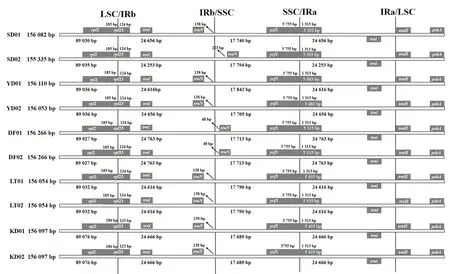
图 2 白花刺续断叶绿体基因组LSC、SSC和IR边缘区的比较Fig. 2 Comparison of LSC, SSC and IR border regions among ten chloroplast genomes in Acanthocalyx alba
利用MISA软件对白花刺续断叶绿体全基因组进行分析,在白花刺续断10条序列中分别检测到70、68、70、70、74、74、70、70、71、71个SSR位点(图3:B)。SSR最丰富的类型为单核苷酸重复,其次是二核苷酸重复、三核苷酸重复、五核苷酸重复、四核苷酸重复和六核苷酸重复。白花刺续断叶绿体基因组中的SSR主要是由A和T组成,其中大部分是以A/T碱基构成的单核苷酸重复,其次是由AT/TA构成的二核苷酸重复(图3:A)。进一步分析表明,大部分SSR位于LSC区域,小部分位于SSC和IR区域(图3:C)。同时,基因组中的SSR大部分分布于基因间区(intergenic spacer, IGS)中,其他少数SSR分布在内含子和蛋白编码区域(coding sequence,CDS)中(图3:D)。

A. SD01叶绿体基因组中不同重复类型SSR出现频率; B. 10个基因组中不同SSR类型的数目; C. 在LSC、SSC和IR区域中出现SSR的频率; D. 在IGS、CDS和内含子中出现SSR的频率。A. Frequency of SSR motifs in different repeat types of SD01 chloroplast genome; B. Number of different SSRs type detected in ten genomes; C. Frequency of identified SSR in LSC, SSC and IR regions; D. Frequency of identified SSR in IGS, CDS and intron.图 3 白花刺续断10条叶绿体基因组的SSR分析Fig. 3 Analysis of simple sequence repeat (SSR) on ten chloroplast genomes in Acanthocalyx alba
2.3 序列差异比较分析
将比对好的5个地区白花刺续断叶绿体基因组进行Sliding window 分析(图4)。结果显示,SSC区域的变异水平最高,IR区域最低。同时,筛选到3条高变异序列,分别位于LSC区(rpoC1)和SSC区(ndhF和rpl32-trnL-UAG)。其中,rpl32-trnL-UAG的变异性最高,其次是ndhF,而rpoC1最低。此外,本研究中以SD01作为参考序列,与其余9条白花刺续断叶绿体基因组进行两两比较分析。结果显示,叶绿体基因组序列中非编码区变异高于蛋白编码区域,单拷贝区(LSC & SSC)变异明显大于反向重复区(IR)。5个地区白花刺续断叶绿体基因组序列整体上高度相似,变异较大的基因有rpoC2、psbC、rrn23和ycf1,其他基因保守程度非常高。基因间区的变异大于基因区,如atpF-atpH、psaB-psaA、psaA-ycf3、trnM-CAU-atpE、psbF-psbE、psbE-petL、rrn5-trnN-ACG、trnR-ACG-trnN-GUU、trnL-UAG-ccsA(图5)。从这些区域中,可开发特异性片段,用于该属种间及种下水平的系统进化与发育研究。
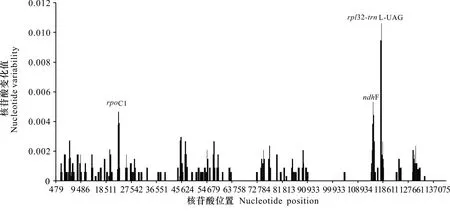
窗口长度为600 bp, 步长为200 bp。X轴. 窗口中点的位置; Y轴. 每个窗口的核苷酸多样性。Window length is 600 bp, step size is 200 bp. X-axis. Position of the midpoint of a window; Y-axis. Nucleotide diversity of each window.图 4 白花刺续断10条叶绿体全基因组的滑动窗口分析Fig. 4 Sliding window analysis of ten chloroplast genomes in Acanthocalyx alba
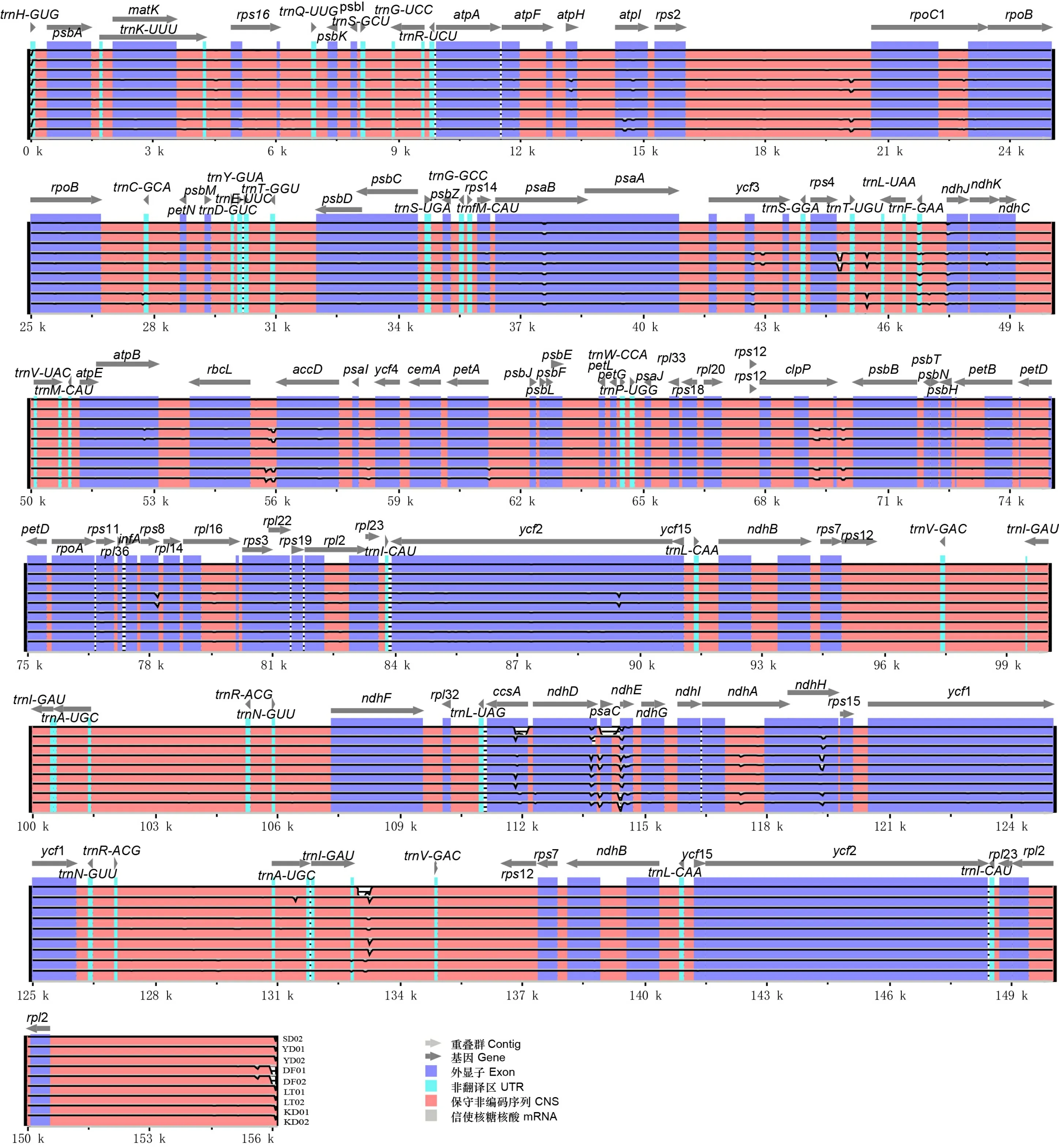
基于VISTA的标识图显示以SD01为参照,对九个白花刺续断进行序列鉴定。VISTA-based identify plot showing sequence identify among nine Acanthocalyx alba using SD01 as a reference.图 5 白花刺续断10条叶绿体基因组的可视化比对Fig. 5 Visualization alignment of ten chloroplast genomes in Acanthocalyx alba
2.4 系统发育分析
本研究利用P-distance种间遗传变异及核苷酸替换比较了10条白花刺续断的叶绿体全基因组进化差异,研究结果表明,P-distance为 0~0.000 7,核苷酸差异值为0~1 515,且大部分序列间地理位置越远, 其相互间P-distance和核苷酸差异值越大(表4)。系统发育分析结果显示,3种方法构建的进化树所反映的不同野生居群之间的进化关系相似(图6),也与遗传距离分析的结果相吻合。在系统发育树中,康定(KD)和道孚(DF)的4个个体最早分化出来,其次是亚丁(YD)和桑堆(SD),最后是理塘(LT)的2个个体。但亚丁(YD)和桑堆(SD)的4个个体不能明显分开。

表 4 白花刺续断个体间遗传距离与核苷酸差异值Table 4 Genetic distances and nucleotide difference values among individuals of Acanthocalyx alba
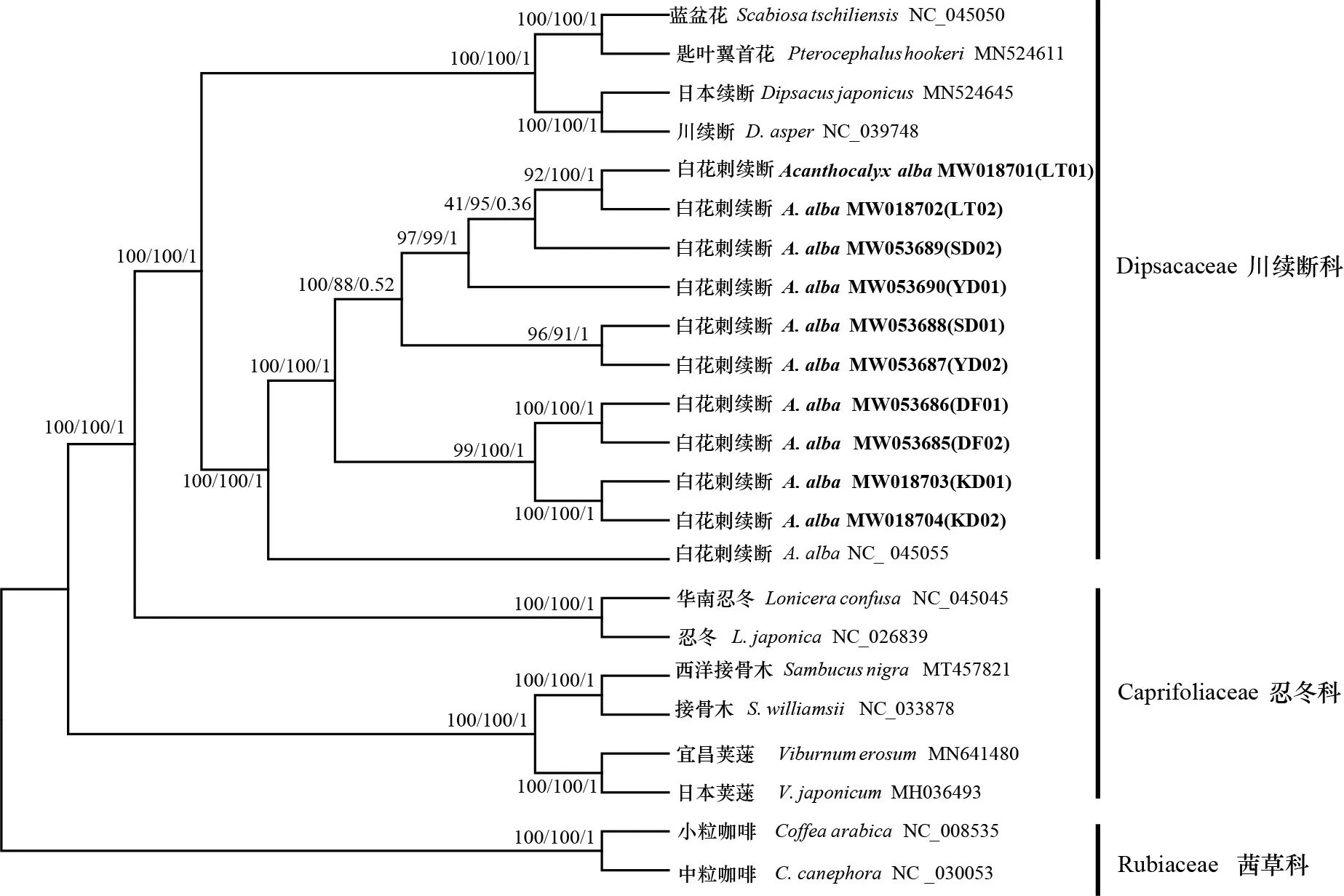
上面的节点数是支持值,左边是MP自展值,中间是ML自展值,右边是贝叶斯后验概率(PP)值。图中加黑部分为本文主要研究对象。Number above nodes are support values with MP bootstrap values on the left, ML bootstrap values in the middle, Bayesian posterior probabilities (PP) values on the right. The black part in the figure is the main research objects of this paper.图 6 利用最大简约法(MP)、最大似然法(ML)和贝叶斯分析法(BI)研究了10个白花刺续断叶绿体全基因组的系统发育关系Fig. 6 Phylogenetic relationship of ten Acanthocalyx alba based on complete chloroplast genome using maximum parsimony (MP), maximum likelihood (ML), and Bayesian analyses (BI) methods
3 讨论与结论
本研究报道了白花刺续断的叶绿体全基因组序列特征,并在居群水平上揭示了其地理遗传结构。不同野生居群的叶绿体基因组所编码的基因类别、数量及排列顺序高度一致。同时个体间具有高度相似的GC含量,单个序列中IRs区序列的GC含量最高。白花刺续断叶绿体基因组中共含有7个假基因,其中5个假基因是川续断科植物所共有的(clpP、accD、ycf2、ycf1、rps18),故推测可能普遍存在川续断科植物假基因现象(Wang et al., 2020)。叶绿体SSR位点是一种高效的分子标记。本研究中,白花刺续断叶绿体全基因组序列的SSRs主要以A/T碱基为主,这与其他被子植物中的情况相似(Guo et al., 2017;Na et al., 2018;Chen et al., 2019)。同时,这也进一步证实了叶绿体SSRs 主要是由polyA和polyT重复所构成,而较少含有C或G串联重复的观点(Kuang et al., 2011)。此外,这些SSRs主要分布在2个单拷贝区,故推测这些高A/T含量的SSRs和分布于IR区的rRNA序列可能是导致叶绿体基因组中GC含量偏低以及各区域碱基含量差异的潜在原因(张明英等,2020)。
IR区和SC区的扩张和收缩被认为是直接影响被子植物叶绿体基因组大小的重要因素(Wang et al., 2017;Song et al., 2019)。本研究表明,不同野生居群个体间叶绿体基因组4个边界均未出现明显的扩张和收缩现象,说明白花刺续断叶绿体基因组IRs区大小高度保守,这也与Wang等(2020)研究结果一致。从叶绿体基因组中发掘的高变片段,不仅可以在物种水平上用于系统发育和物种鉴定研究,也可以在居群水平上提供丰富的遗传信息,从而揭示物种的居群动态与进化历史等。Fatemeh等(2018)基于rpl32-trnL-UAG对滇紫草属(Onosma)物种进行系统发育分析和分化时间估计;Nahla等(2020)采用rpoC1对苜蓿属(Medicago)植物进行亲缘关系分析;Chen等(2020)基于叶绿体基因组对贝母属(Fritillaria)植物进行研究,结果发现筛选出的ycf1和psbM-psbD可作为特定条形码用于贝母属植物物种鉴定。这些研究工作也进一步证实高变片段在物种进化及鉴定等方面具有特殊作用。本研究中,筛选出3个高变片段(rpoC1、ndhF和rpl32-trnL-UAG),可用于刺续断属内种间系统发育及种内群体遗传学研究。
传统上,常用叶绿体基因片段来研究物种的群体遗传结构和谱系进化关系,但因叶绿体片段多态位点不足而作用有限(Zhang et al., 2019;Zhang et al., 2020;刘家奇等,2021)。与之相比,叶绿体全基因组具有极为丰富的遗传变异,为复杂植物类群的遗传进化研究提供有效手段。Wang等(2020)基于美国山核桃(Caryaillinoinensis)两个不同居群间叶绿体基因组核苷酸差异性,揭示了该物种居群水平的遗传多样性。本研究中,白花刺续断5个野生居群间具有较为明显的遗传结构,个体间的遗传距离、核苷酸差异值与地理距离之间呈较好的相关性。这也与系统发育树所揭示的进化关系相吻合。值得注意的是,桑堆(SD)和亚丁(YD)的四个个体没有形成独立分支,这可能是两个居群间地理距离较近引起相对频繁的基因流所致。这一结果也说明,与核基因组相比,叶绿体基因组进化较慢及单亲遗传的特性,其作用也有明显的局限性。
综上所述, 叶绿体全基因组序列具有极为丰富的遗传信息,可为复杂植物类群及种下居群水平上的群体遗传及谱系进化研究提供有效手段。但由于二代测序的价格仍旧较为高昂,本文中居群样本量较少,本研究结果的科学性尚有不足。因此,叶绿体基因组能否作为传统的分子片段或标记的技术补充,需要更多研究工作来验证。此外,将叶绿体全基因组用于群体遗传学分析的数据分析方法也有待于进一步完善。
