基于MOF 衍生制备Pd-CeO2 催化剂及其催化氧化性能
2022-11-15叶菁睿何光裕陈海群
叶菁睿,何光裕,陈海群
1.常州大学石油化工学院,江苏 常州 213164;
2.浙江大学化学工程与生物工程学院,浙江 杭州 310027
一氧化碳是一种无色无味的有毒气体,对人体健康和自然环境都有不良影响,当环境中CO 浓度超过400 mg/kg 即有可能危及生命,因此有必要开展低浓度CO 的消除研究。此外,CO 经常被用作分子探针,用以研究催化剂的物理化学特性、吸脱附过程和反应机理,而且CO 氧化反应耗费低,易于实施,常被视作模型反应,因此CO 催化氧化反应吸引了众多研究者的注意[1-3]。该反应所用的催化剂可分为非贵金属催化剂和贵金属催化剂。非贵金属催化剂价格便宜,但催化活性普遍不高,对水汽条件敏感;贵金属催化剂中,Pd 基催化剂因其较为理想的催化活性和稳定性而受到青睐[2]。Pd 基催化剂的常用载体包括还原性载体(如CeO2,TiO2和Co3O4等)和非还原性载体(如C,SiO2和MgO等)两种类型,其中CeO2存在Ce3+和Ce4+之间的循环变换,表现出高储氧能力和高氧流动性,被视作较为理想的Pd 基催化剂载体[4-5]。
负载型Pd-CeO2催化剂可采用浸渍法、共沉淀法、化学还原法和离子交换法等方法制备,另外,金属有机框架(MOFs)辅助制备负载型催化剂的方法被越来越多的研究者采纳。金属有机框架,也称为多孔配位聚合物,是由金属离子或团簇与有机配体自组装而成的类沸石材料[6-7]。金属中心可为3d过渡金属、3p金属或者镧系元素,灵活的组成结构为MOFs 的应用提供了无限的可能性。此外,MOFs 通常可热解转变为氧化物,因此可作为可牺牲模板制备负载型催化剂。Luo 等[8]将Pt 负载在铈基MOF 和碳纳米管上,并热解制备Pt/CeOx/C 催化剂,发现MOF 结构可以在热处理过程中防止Pt团聚。本工作以铈离子作为金属节点,对苯二甲酸作为有机配体,组装铈基MOF 材料,并在组装过程中引入Pd,得到负载Pd 的铈基MOF(Pd-CeMOF),通过后续热解操作制备Pd-CeO2催化剂。通过对Pd-CeMOF 及其衍生物Pd-CeO2样品的表征和活性评价,探究这一制备方法的优势以及在CO 氧化反应中的催化性能。
1 实验部分
1.1 催化剂制备
取一定质量的氯化钯加入HCl 水溶液中,溶解稀释后定容为2.0 mmol/L 氯钯酸溶液。将48 mL氯钯酸溶液、45 mL 去离子水、30 mL 乙醇和0.40 g 聚乙烯吡咯烷酮(PVP)加入到250 mL 圆底烧瓶中,在110 ℃下搅拌回流3 h,再将其旋转蒸发后得到PVP 封端的Pd 纳米颗粒,记作Pd-PVP[9]。
取0.08 mmol 的Pd-PVP(Pd 负载量为0.5%质量分数)溶于60 mL 混合溶剂(乙醇与水的体积比为1:1),依次加入0.97 g 对苯二甲酸(1,4-H2BDC)与2.52 g 六水合硝酸亚铈,在室温下搅拌10 min后形成均匀的体系。将其装入具有聚四氟乙烯内衬的金属反应釜中,160 ℃下晶化24 h,所得混合物经过离心分离,固体分别用水和乙醇洗涤数次,并在60 ℃下干燥过夜,得到负载Pd 的铈基MOF,记为Pd-CeMOF。将上述制得的Pd-CeMOF在静态空气中于500 ℃下焙烧2 h(升温速率为3 ℃/min),得到Pd-CeO2催化剂。
未负载Pd 的CeMOF 和CeO2样品通过类似的水热法合成。将0.36 g PVP 溶于60 mL 混合溶剂(乙醇与水的体积比为1:1),依次加入0.97 g 对苯二甲酸与2.52 g 六水合硝酸亚铈,室温下搅拌10 min后160 ℃下晶化24 h。离心分离洗涤后于60 ℃下干燥得到CeMOF 样品。将CeMOF 在静态空气中于500 ℃下焙烧2 h(升温速率为3 ℃/min)后,得到CeO2样品。
1.2 催化剂表征
红外光谱(FTIR)是由Nicolet 5700 型光谱仪表征。热重分析(TGA)是在Mettler Toledo 公司的TGA/DSC 3+仪器上进行。粉末X 射线衍射(XRD)是由X´Pert3 X 射线衍射仪表征,使用Cu-Kα入射源,工作电压为40 kV,工作电流为40 mA,扫描2θ为5°~100°,扫描步长为0.02°。电感耦合等离子体发射光谱(ICP)是由Optima 5300DV 光谱仪测得,其中CeO2和Pd-CeO2样品测试前需经由浓硝酸和过氧化氢加热溶解,CeMOF 和Pd-CeMOF 样品测试前需经由硝酸在水热釜中加热溶解。X 射线光电子能谱(XPS)是由Escalab 250Xi 型仪器测得,并经由标准C 1s 峰位置(284.6 eV)校正。透射电子显微镜(TEM)图像和能谱分析(EDS)采用配备Oxford X-MAXn65 T 模块的HT-7700 型电镜表征样品的形貌特征。氢气程序升温还原(H2-TPR)是由AutoChemⅡ 2920 型化学吸附仪表征,升温速率为10 ℃/min。拉曼光谱(Raman)是由LabRAM HR800 型光谱仪表征,采用514 nm 的光源。是由配备Oxford X-MAXn65 T 模块的HT-7700 型电镜表征。
1.3 催化剂的活性评价
催化剂的活性评价在固定床反应器中进行。不锈钢反应管的内径为8 mm,下端填充石英棉,恒温区的长度为40 mm,装填粒径为0.3~0.45 mm(40~60 目)的催化剂0.1 g。以一氧化碳体积分数为1%,氧气体积分数为1.55%的混合气体(稀释气体为氦气)进料,空速为15 L/(gcat·h)。反应在常压下进行,升温速率为3 ℃/min。通过在线色谱(奇阳GC 9860)对反应产物进行监测,所用色谱柱为TDX-01,所用检测器为热导检测器(TCD)。
2 结果与讨论
未负载和负载Pd 的铈基金属有机框架的红外谱图如图1 所示。由图1 可见,两种样品的红外谱图中均不存在非离子态羧基基团的特征峰(如C=O在1 686~1 720 cm-1处和—OH 在3 090 cm-1处的特征峰),并且都存在离子态COO—基团的特征峰,也就是位于1 479~1 608 cm-1处的非对称伸缩振动峰和位于1 391~1 450 cm-1处的对称伸缩振动峰[10-11]。这些离子态COO—基团特征峰的出现表明Ce 离子与对苯二甲酸间之间存在配位作用。3 450 cm-1处特征峰是由于水分子中的强OH 伸缩峰而出现的,这表明水分子也在铈基MOF 中具有配体作用。由红外分析确定,以铈离子为金属节点,以对苯二甲酸为有机配体,可以组装成铈基金属有机框架材料,而在组装过程中加入Pd 对金属有机框架材料的形成和结构没有影响。

图1 催化剂样品的红外图谱Fig.1 FTIR spectra of catalyst samples
催化剂样品的XRD 谱图如图2 所示。图2(a)中位于9.7°,15.1°和18.7°的特征峰与文献报道中以铈离子为金属节点,对苯二甲酸(1,4-BDC)为有机配体,化学式为Ce(1,4-BDC)1.5(H2O)2的MOF材料峰位置一致[12]。在制备过程中引入Pd-PVP 后,Pd-CeMOF 样品的XRD 谱图并未出现金属Pd0(40.1°)或者PdO(33.8°)的信号,这可能是因为Pd 负载量较低,或者是被CeMOF 的信号所掩盖[13]。对铈基MOF 材料热解后的衍生物也进行了XRD 表征,结果如图2(b)所示。图2(b)谱图中的衍射峰归属为面心立方型CeO2萤石结构(JCPDS #34-0394),所有CeMOF 的特征峰都已消失,说明铈基MOF 材料在热处理过程中已经完全热分解为CeO2。由于Pd 含量较低,CeO2和Pd-CeO2样品的XRD 图谱中也不存在Pd0或者PdO 的信号,这表明Pd 颗粒在铈基MOF 热解过程中并未发生严重烧结。

图2 催化剂样品的XRD 图谱Fig.2 XRD patterns of catalyst samples
CeMOF 和Pd-CeMOF 的热重曲线如图3 所示。由图3 可知,两种催化剂的热失重曲线变化不大,这说明Pd 的引入对CeMOF 的热稳定性影响很小。图3 中的热重曲线主要存在两个明显的失重过程:温度为100~250 ℃时的失重主要是由于作为配体的水分子发生脱离,通过XRD 表征确定铈基MOF的组成是Ce(1,4-BDC)1.5(H2O)2,这一阶段约10%有质量损失,与Ce(1,4-BDC)1.5(H2O)2脱去两个水分子的质量比例相符;而在400 ℃左右出现的迅速失重阶段则是由于有机配体发生完全分解,铈基MOF 转化为 CeO2[14],整个分解过程中质量损失约为47%~48%,与Ce(1,4-BDC)1.5(H2O)2转化为CeO2的失重比例基本相符。

图3 催化剂样品的热重分析Fig.3 Thermogravimetric analysis cures of catalyst samples
图4(a)为TEM 表征的催化剂微观形貌。由图4(a)可见,CeMOF 为规整圆球状,直径约为100~800 nm,圆球的边缘粗糙;负载上Pd 后的样品Pd-CeMOF,其形貌有所变化,变为非正圆的近似球状,其内外存在明显的衬度差别,球体边缘较厚,中间较薄,表明Pd-PVP 的引入使得铈基MOF 的生长发生改变,形貌转变为较薄的近球体,而且Pd-CeMOF 表面粗糙,由颗粒组成。由于CeMOF 材料较为致密,无法通过TEM 直接观察到其上的Pd,进一步通过EDS 分析确认了Pd 的成功负载,如图4(b)所示,Pd 信号集中于球体附近,表明Pd-CeMOF 上Pd 的分散较为均匀,表明将Pd 引入MOF 后进行热解是一种制备负载型催化剂的有效方法,可以在制备过程中防止贵金属团聚。由图4(a)所示的衍生物CeO2和Pd-CeO2样品的形貌可知,其与前驱体相比基本没有变化;CeO2中组成球体的颗粒堆积得较为致密,Pd-CeO2中组成近球体的颗粒堆积得较为稀疏。

图4 催化剂样品的TEM 图片(a)和Pd-CeMOF 的EDS 元素扫描图谱(b)Fig.4 TEM images of catalyst samples (a) and EDS elemental mappings over Pd-CeMOF (b)
通过ICP 表征以确定Pd-CeMOF 和Pd-CeO2中的Pd 含量。Pd-CeMOF 中Pd 的质量分数为0.49%,与理论负载量0.50%较为接近,说明Pd-PVP 的预合成可在铈离子和对苯二甲酸的组装过程中有效防止贵金属Pd 的流失;Pd-CeO2中Pd 的质量分数为1.04%。由XPS 测得Pd-CeMOF 和Pd-CeO2表面Pd 含量分别为0.11%和0.58%,与ICP 相比都较低,这说明在Pd-CeMOF 中,Pd 更多位于铈基MOF框架内部,而在其衍生物中,Pd 主要位于由CeO2颗粒组成的孔中。
图5 是催化剂的XPS 表征及分峰结果。其中由Ce3+的3d能级激发的特征峰位于902.8,899.7,884.4 和881.7 eV 处,由Ce4+的3d能级激发的特征峰则是位于917.3,907.9,901.3,898.9,889.1 和882.8 eV 处[15]。CeMOF 和Pd-CeMOF 中同时存在Ce3+和Ce4+离子。在其衍生物CeO2和Pd-CeO2催化剂中,Ce4+浓度有所增大,Ce 元素从以Ce3+形式为主转变成了以Ce4+为主。Pd-CeMOF 中Pd 以两种形态存在,即Pd0和PdO,分别激发位于335.2 eV 和336.9 eV 附近的3d5/2特征峰[16-17]。且Pd 在Pd-CeMOF 中主要是以金属Pd0的形式存在,小部分被氧化成PdO。对衍生物Pd-CeO2的曲线进行分峰后,发现主要存在两种Pd 物种,即由PdO 激发的336.8 eV 处特征峰以及位于337.8 eV 处的特征峰。位于337.8 eV 处3d5/2特征峰对应于与CeO2产生相互作用的高离子态Pd 物种(Pdn+)[18-19]。表明在铈基MOF 热解过程中,大部分Pd 被氧化成PdO,且在这一过程中,Pd 与CeO2出现相互作用。
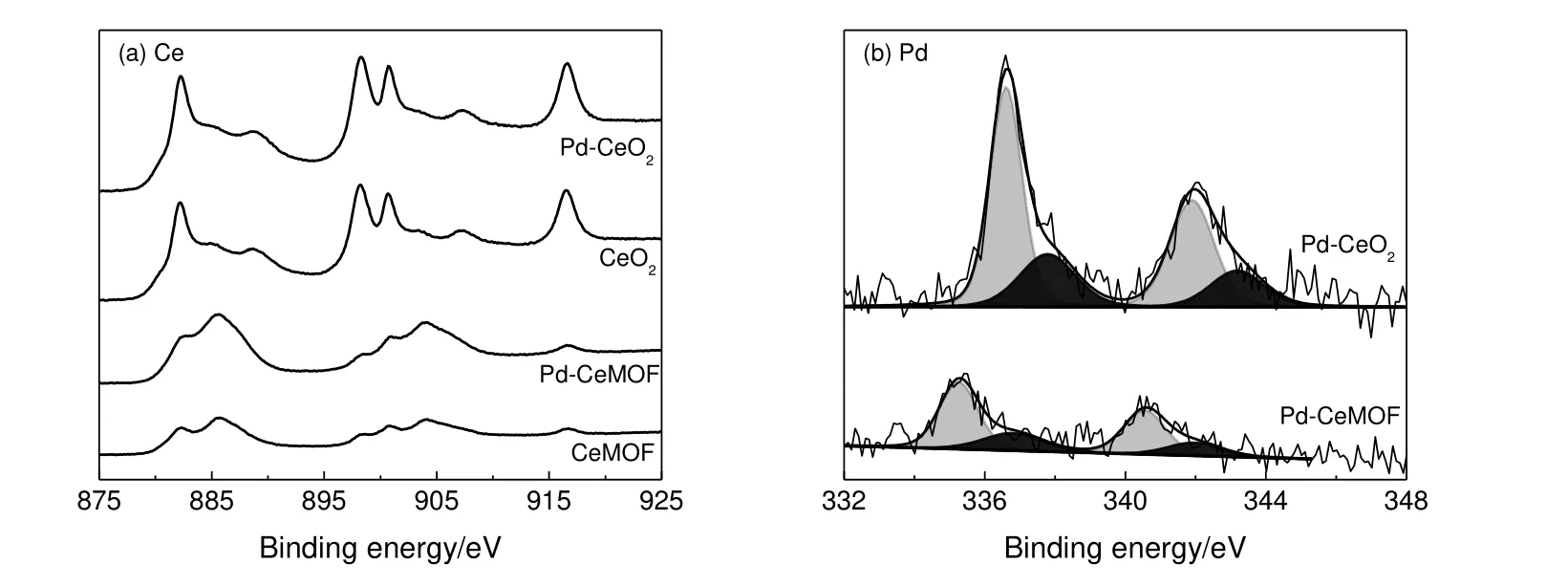
图5 Ce 3d XPS 谱图和Pd 3d XPS 图谱Fig.5 Ce 3d XPS spectra and Pd 3d XPS spectra
在此基础上,通过H2-TPR 表征继续研究表面Pd 物种状态。如图6 所示,CeO2中主要有两个还原峰。高温处(800 ℃左右)的氢气消耗峰对应于CeO2体相氧的还原,450 ℃处消耗峰对应于CeO2表面氧的还原[20]。Pd-CeO2中CeO2体相氧和表面氧的还原性能与未负载的样品类似,不同之处主要在于低温(200 ℃以下)还原峰。低温还原峰涉及Pd 物种的还原,其峰面积所反映的H2消耗量明显高于PdO 还原所需要的量,可以推测其中存在CeO2的还原行为[21]。在Pd-CeO2的还原曲线中,73 ℃处的H2消耗峰对应于与CeO2产生相互作用的Pdn+物种。此外,61 ℃处的负峰表示有H2生成,这是由于Pd 与H2形成PdHx后,温度升高后再分解生成H2[22]。PdO 物种的还原在温度较低处发生,因此未能被H2-TPR 曲线记录下来。

图6 CeO2 和Pd-CeO2 的H2-TPR 图谱Fig.6 H2-TPR profiles of CeO2 and Pd-CeO2 samples
图7 是CeO2和Pd-CeO2的拉曼光谱谱图。CeO2和Pd-CeO2中主峰位于455 cm-1处,对应于立方萤石结构CeO2的三重简并态F2g振动模式,1 174~1 180 cm-1处峰对应于CeO2中二阶振动模式[23]。590 cm-1处肩峰对应于氧空穴的缺陷诱导D 振动模式,240 cm-1处峰是氧空穴引发的二阶横向声学振动模式[24],这两处峰的强度都与样品表面的缺陷浓度有关。与载体CeO2相比,Pd-CeO2催化剂中Pd与Ce 之间存在相互作用,从而引起电子的再分配,为维持电中性而产生了缺陷结构,因此Pd-CeO2表面的氧空穴浓度有所提升。此外,Pd-CeO2催化剂在651 cm-1处出现特征峰,对应于PdO 的振动模式,与XPS 及H2-TPR 结果相符[21]。

图7 CeO2 和Pd-CeO2 的拉曼图谱Fig.7 Raman spectra of CeO2 and Pd-CeO2 samples
CeO2,Pd-CeO2及其前驱体Pd-CeMOF 的CO氧化反应活性考评曲线如图8 所示。由图8 可知,CeO2,Pd-CeO2和Pd-CeMOF 在CO 转化率达到50%时的温度分别为261,131 和224 ℃。CO 转化率达到90%时的温度分别为308,162 和229 ℃。Pd-CeO2的催化活性显著高于纯载体及其前驱体,特别是CeO2和Pd-CeMOF 在低温区(小于180 ℃)的活性均可忽略不计。这说明在MOF 中引入Pd 后热解是一种有效制备负载型Pd 催化剂的方法。Pd 是CO 氧化反应中的主要活性成分,尤其是针对低温阶段,可大幅提高催化活性。而同Pd-CeMOF 相比,Pd-CeO2中活性的提高是因为衍生物具有更高的缺陷浓度,以及Pd 与CeO2之间的相互作用。虽然Pd 基催化剂在CO 氧化反应中的反应机理仍存在争议,但是研究者普遍认可活性金属与载体之间的界面在这一反应中具有重要作用[17]。对于有界面参与的Mars-van Krevelen 机理来说,通常认为是在界面Pd 原子处发生反应,由Pd 吸附CO,并与活化晶格氧之间发生反应,之后由O2吸附解离补充氧空穴[25]。由拉曼表征可以发现Pd-CeO2的氧空穴浓度相比Pd-CeMOF 有所提高,而由XPS 和H2-TPR 共同揭示了Pd-CeO2催化剂中Pd-Ce 相互作用和Pdn+物种的存在。结合Mars-van Krevelen 机理来看,Pd-CeO2催化剂中Pd-Ce 相互作用增大并强化了界面,从而可以提供更多活性位点,而表面缺陷浓度增大使得O2流动性增强,可以更快为反应提供吸附解离氧。因此,Pd-CeO2催化剂表现出了更高的CO 催化氧化活性。

图8 CO 氧化反应活性曲线Fig.8 Light-off curves of CO oxidation
3 结 论
基于MOF 衍生制备了负载型Pd-CeO2催化剂,通过表征发现,MOF 在制备过程中有效防止贵金属的流失和团聚,MOF 衍生过程可以促进Pd 与CeO2形成金属-载体相互作用,且催化剂中氧空穴浓度也得以提高。通过反应考评发现Pd-CeO2催化剂可在131 ℃下达到50%转化率,在162 ℃下达到90% 转化率,归因于增强的Pd-Ce 相互作用为反应提供更多活性位点,且增大的表面缺陷浓度使得O2流动性增强。
