LilrB2蛋白与Aβ(16-21)双链的结合*
2022-11-15吴汶泽李晓毅
吴汶泽,李晓毅
(中国科学院大学材料科学与光电技术学院 材料科学与光电技术中心, 北京 100049)
阿尔茨海默症(Alzheimer’s disease,AD)是一类神经退行性疾病,是威胁人类身体健康的重大隐患之一。有报告指出,全球约有4 000万人罹患阿尔兹海默症,而这个数字预计将每20年增加一倍。Aβ1-42是阿尔兹海默症患者的患病标志物之一,一方面,Aβ1-42寡聚态对神经细胞具有毒性,可诱导神经细胞凋亡。另一方面,Aβ1-42寡聚态可与神经细胞上的某些受体结合,导致其下游通路上突触损失,造成认知障碍[1-2]。针对Aβ1-42寡聚态所带来的危害,目前有2种抑制思路:一是以Aβ1-42寡聚态为靶点,设计药物抑制Aβ1-42寡聚态的活性,降低Aβ1-42寡聚态对人体造成的影响;二是以Aβ1-42寡聚态的受体蛋白为靶点,抑制受体蛋白的活性从而达到缓解AD症状的效果,而后者的优势在于在AD发病的早期就可以看到明显的效果[3]。
基于上述想法,寻找Aβ1-42寡聚态的靶向蛋白是一个重要问题。Taeho等[4]通过研究发现小鼠上PirB蛋白是Aβ1-42受体之一,PirB与Aβ1-42寡聚态的结合会使得下游通路上的cofilin(AD另一种患病标志物)量增大,小鼠有明显的认知水平缺失,通过比较人类与小鼠的同源基因,发现人神经细胞上lilrB2蛋白会与Aβ1-42寡聚态结合,且造成下游通路上的cofilin量增大。屏蔽lilrB2蛋白与Aβ1-42寡聚态的结合可作为一种药物靶向来设计针对AD的抑制剂。更进一步,Qin等[3]研究了lilrB2与Aβ1-42的结合,比较发现,Aβ16-21双分子链可代表Aβ1-42寡聚态,作为与lilrB2蛋白结合的核心抗原基,研究lilrB2与Aβ16-21双分子链的结合机理有助于设计相应的抑制剂。而目前关于lilrB2蛋白抑制剂先导化合物的研究报道还较少,研究lilrB2与其天然配体的结合模型对寻找和设计先导化合物有重要意义。
计算机辅助药物设计(computer-aided drug design,CADD)是随着分子生物学,计算机技术发展而应运而生的一种研究手段,由于其快速,高效以及可程序化的特点,计算机辅助渗透到新药研发的各个方面。与传统药物设计相比,CADD可显著提高药物研发的成功率,降低研发成本,缩短研发周期[5-6]。基于受体结构的药物分子设计是CADD应用的方法之一,即在已知受体三维结构的基础上,通过分子对接以及分子动力学的方法研究受体与配体相互作用的方式和特点,从而为设计受体蛋白抑制剂提供分子层面的支持[7-10]。
目前,关于lilrB2蛋白抑制剂先导化合物的研究报道较少,本文以lilrB2蛋白为靶点,通过分子对接和分子动力学的方法,研究lilrB2蛋白与其配体Aβ16-21双分子链之间结合的方式以及相互作用的热点氨基酸残基,为lilrB2蛋白抑制剂先导化合物的寻找和设计提供分子层面的数据支持。
1 材料和方法
1.1 模型搭建
本文所使用的lilrB2蛋白受体(PDB ID 2GW5)和Aβ16-21双分子链(PDB ID 3OW9)的结构均来源于蛋白质数据库RCSB PDB。LilrB2蛋白包含184个氨基酸残基,形成2个结构域,2个结构域向外伸展,如图1所示。
图1 lilrB2三维结构图(飘带模型)
Aβ16-21双分子链有12个氨基酸残基,双链呈反平行结构,每条链上的氨基酸相同,结构如图2所示。

图2 Aβ16-21双分子链三维结构图(棍状模型)
从蛋白数据库获取晶体结构后,首先利用分子动力学优化其结构。lilrB2蛋白在溶液环境中优化90 ns,取平衡轨迹中不同的2帧结构(均方回转半径分别为1.96和2.02 nm)用于对接。Aβ16-21双分子链在溶液环境中优化50 ns,取平衡轨迹中1帧结构用于对接。将lilrB2蛋白和Aβ16-21双分子链进行分子对接,所得结果如图3所示。
图3 利用分子对接软件得到的对接复合物结构图
由2帧lilrB2结构和1帧Aβ16-21双分子链,得到3个不同结合方式的复合物结构。为探明lilrB2蛋白和Aβ16-21双分子链结合方式的特点以及结合时重要的氨基酸残基,分别以3个复合物结构为起始构象,在溶液环境下进行分子动力学模拟,分别用P1,P2,P3表示。P1和P2进行90 ns的平衡模拟,P3进行80 ns的平衡模拟。
1.2 分子对接和分子动力学模拟
分子对接所使用的软件为autodock 4.2[11],对接方式为刚性对接。分子动力学模型搭建使用的软件是VMD[12],分子动力学运行所使用的软件是NAMD 2.13软件包[13],采用CHARMM27[14]力场,溶液环境所用水分子模型为TIP3P[15],使用周期性边界条件,体系到水盒子边界距离设定为1.5 nm,体系中添加Na+和Cl-,浓度0.15 mol/L。模拟是在NPT系综下进行的,压强1 atm,温度300 K。范德华相互作用阈值设置为1.2 nm。采用粒子网格(PME)方法处理静电相互作用。所有模拟积分步长均为1 fs。本文数据均使用origin 9.1软件作图。
1.3 氢键分析
氢键是分析复合物结合稳定性的重要指标。本文使用VMD HBonds Plugin 1.2进行氢键的统计和分析,所采用的距离阈值为0.35 nm,角度阈值为30°。
2 结果与讨论
2.1 均方根偏差
通过分子对接和分子动力学模拟,得到了以不同构象为起始点的3组轨迹。对这3组轨迹进行数据整理,首先,计算得到这3组轨迹的均方根偏移(root-mean-square deviation,RMSD)值随时间变化的图像,如图4所示。RMSD数据一方面可以反映在模拟过程中复合物结构是否有大的起伏,观察受体蛋白和配体结合方式在哪个时间段上发生了变化。另一方面,RMSD是反映体系状态是否稳定的一个重要指标。
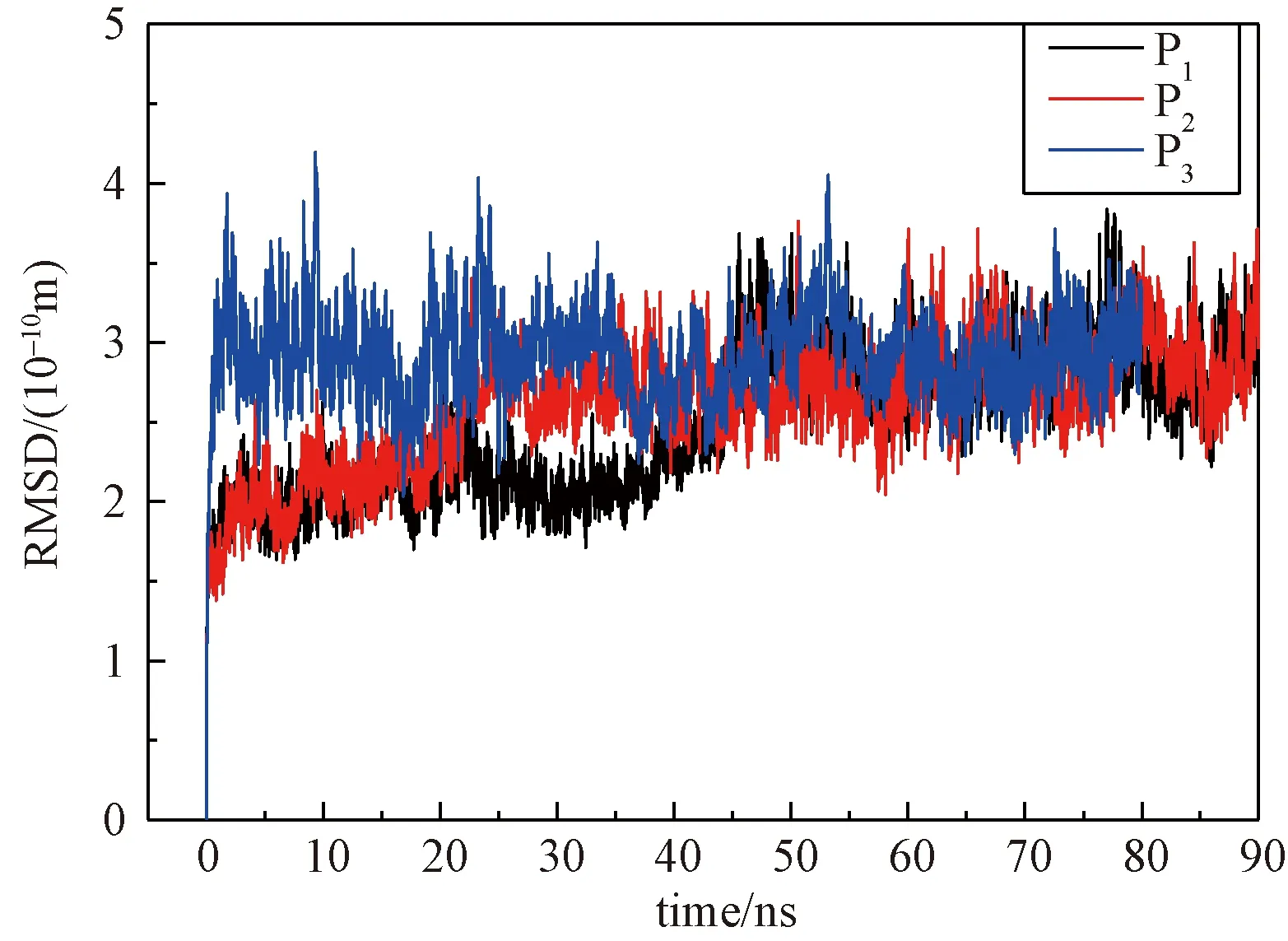
图4 RMSD随时间的变化曲线
从图4可以看到,P1体系在40~50 ns的阶段RMSD值有一个明显的波动,P2在20~30 ns阶段RMSD值有一个明显的波动,而P3的RMSD值一直在0.3 nm附近变动。分析发现,这种波动是由于lilrB2蛋白和Aβ16-21双分子链的结合方式发生了改变。也就是说,lilrB2蛋白和Aβ16-21双分子链的结合倾向了一种更稳定的状态。这种状态正是我们所要探究的,所以以3个不同构象作为起始点进行模拟,以期找到lilrB2蛋白和Aβ16-21双分子链结合状态的共通点。从图4中还可以看到,P1,P2,P3这3组体系的RMSD值最后都稳定在0.3 nm附近,3组体系均已达到平衡状态。
2.2 氢键占有率
对P1体系30~40 ns和50~60 ns时间段做氢键占有率的分析,选取了排名前3的数据来分析,数据见表1。
这里的氢键是通过残基间的距离和角度来判定的,也就是依据残基间的几何特征来判定氢键是否存在,所有在设定阈值内的残基相互作用都被判定为氢键。氢键占有率是指在所截取的时间段内,该氢键出现的频率。
从表1数据可以看出,从30~40 ns阶段到50~60 ns阶段,ARG77-ALA12的氢键占有率由41.20%降到了11.40%,在50~60 ns阶段,氢键占有率排第1的是ASP23-LYS7,为17.10%。残基对之间的氢键占有率变化暗示着P1体系中受体和配体之间的结合方式发生了一些变化。
对P2体系10~20 ns和30~40 ns时间段做氢键占有率的分析,数据见表2。
由表2中数据可见,在10~20 ns时间段内,ARG77-ALA12的氢键占有率排在第1,为69.10%,而在30~40 ns时间段,ASP23-LYS7的氢键占有率居首位,为90.70%,这暗示着P2体系中受体和配体之间的结合方式也发生了变化。
结合表1和表2,lilrB2蛋白和Aβ16-21双分子链之间的结合方式发生改变,且在改变之后,ASP23和LYS7的氢键占有率一直排在首位,考虑到LYS是碱性氨基酸,带1个正电荷,ASP是酸性氨基酸,带1个负电荷,ASP和LYS之间容易形成较强的离子相互作用,所以推测ASP23和LYS7之间的相互作用可能是使得lilrB2蛋白和Aβ16-21双分子链之间结合形成更稳定状态的因素之一。

表1 P1中相互作用残基的氢键占有率

表2 P2中相互作用残基的氢键占有率
为进一步研究这一种状态的结合方式,对3组轨迹的最后10 ns数据进行氢键分析,依旧选取氢键占有率排名前三的数据,数据见表3。
根据表3中数据,P1体系中氢键占有率最高的是ASP23-LYS7,为43.00%;其次是CYS142-VAL3,为16.40%;排第3的是GLU140-PHE5,占有率为6.50%。P2体系中氢键占有率最高的是ASP23-LYS7,为83.70%;排第2的是SER24-ALA6,为32.20%;排第3的是ASP93-LYS1,占有率是8.90%。P3体系中氢键占有率最高的是ASP23-LYS1;排第2的是CYS138-LYS1,为33.90%;CYS142-PHE10的氢键占有率为22.00%,排第3。结合来看,lilrB2蛋白上的ASP23残基与Aβ16-21双分子链上的LYS残基之间的相互作用对lilrB2蛋白和Aβ16-21双分子链的结合起到了很大的稳定作用。而对于其它氨基酸残基,疏水作用也对复合物的稳定性起到了重要作用,在不同结合状态下,有CYS142对Aβ16-21支链上VAL的作用,SER24对Aβ16-21支链上ALA的作用,CYS142对Aβ16-21支链上PHE的作用。也就是说,lilrB2与Aβ16-21双分子链形成的复合物具有较好的稳定性,且维持着一种动态平衡,在这种动态平衡中,ASP23与Aβ16-21支链上LYS残基的离子相互作用对复合物的结合有着重要的影响,而在受体和配体不同结合状态下,lilrB2蛋白上CYS142和SER24残基与Aβ16-21支链上VAL、PHE、ALA残基的疏水作用也有利于lilrB2蛋白和Aβ16-21双分子链结合。
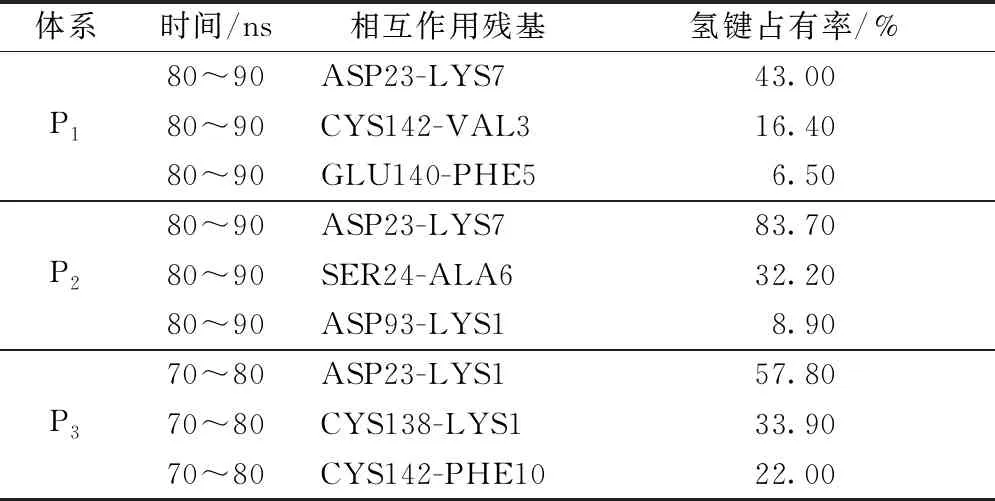
表3 3组体系中相互作用残基的氢键占有率
3 结论
本文通过分子对接和分子动力学模拟的方法研究lilrB2蛋白和Aβ16-21双分子链的结合模型以及在结合中起重要作用的氨基酸残基。通过设置3个不同构象作为分子模拟的起始点,得到3组不同的模拟轨迹,分析发现在受体和配体结合过程中lilrB2蛋白上的ASP23残基与Aβ16-21支链上的LYS残基有较强的相互作用,对受体和配体结合起到了重要的影响。在受体和配体不同结合状态下,CYS142和SER24残基与Aβ16-21支链上VAL、PHE、ALA残基的疏水作用也有利于lilrB2蛋白和Aβ16-21双分子链结合。
