高效液相色谱法检测对乙酰氨基酚片中有关物质*
2022-11-11帅海涛章泽恒黄致远
肖 江,王 辉,帅海涛,章泽恒,黄致远
(1.湖南省药用辅料工程技术研究中心有限公司,湖南 长沙 410331;2.国家药用辅料工程技术研究中心,湖南 长沙 410331;3.湖南省药品审评认证与不良反应监测中心,湖南 长沙 410013)
对乙酰氨基酚又称扑热息痛,化学名为4'-羟基乙酰苯胺,为乙酰苯胺类解热镇痛药,临床广泛用于治疗各种轻、中度疼痛及儿童的发热疾病[1-4]。对乙酰氨基酚于1878年在约翰·霍普金斯大学首次合成,1951年在美国获批上市,1955年成为非处方药,并成为世界上应用最广的解热镇痛药。我国最早于1960年开始生产[5]。目前,2020年版《中国药典》[6]、《欧洲药典》10.0版[7]和《美国药典》40版[8]均收载了对乙酰氨基酚的有关物质检查方法,除对氨基酚与对氯苯乙酰胺采用外标法测定外,其他杂质均采用不加校正因子的主成分自身对照法测定。根据对乙酰氨基酚的合成工艺推测,原料药中潜在的杂质有杂质A-I和杂质L-N,均含有基因毒性杂质的潜在结构[9]。杂质L-N为《欧洲药典》10.0版中新增杂质[8,10],杂质L由苯酚氯代产生的副产物杂质C与对乙酰氨基酚聚合而成,杂质M和杂质N分别为对氨基酚和对乙酰氨基酚在酸性高温条件下形成的聚合物。对氨基酚和对氯苯乙酰胺均为毒性杂质,具有潜在的基因毒性[11-12]。目前,关于乙酰氨基酚原料药及制剂有关物质的研究多为对氨基酚、对氯苯乙酰胺、杂质A、杂质B、杂质D、杂质F,对大部分潜在杂质未开展系统性研究,且对国内制剂的质量也未进行考察[11-17]。本研究中在各国药典方法的基础上建立了一种带校正因子的主成分自身对照法,对国内多个厂家的对乙酰氨基酚片与参比制剂进行了杂质对比研究,并根据2020年版《中国药典(四部)》[18]和国际人用药品注册技术协调会(ICH)指导原则Validation of Analytical Procedures:Text and Methodology Q2(R1)进行了全面的方法学考察。现报道如下。
1 仪器与试药
1.1 仪器
Agilent1260型高效液相色谱仪(美国Agilent公司),配有紫外检测器;XS-205型电子天平(瑞士梅特勒-托利多公司,精度为十万分之一);KQ5200B型超声波清洗器(昆山市超声仪器有限公司,功率为200W,频率为40kHz);Milli-Q型超纯水制水机(德国默克密理博公司)。
1.2 试药
对乙酰氨基酚片(12批,规格均为每片0.5 g),样品信息及生产厂家见表1;有关物质对照品信息见表2(杂质A-I和杂质L-N由TLC Pharmaceutical Standards公司提供;对氨基酚,对氯苯乙酰胺,对乙酰氨基酚由中国食品药品检定研究院提供);冰醋酸(分析纯,天津市大茂化学试剂厂);甲醇(色谱纯,德国默克公司);水为超纯水。

表1 对乙酰氨基酚片样品信息及生产厂家Tab.1 Information and manufacturers of Paracetamol Tablets samples
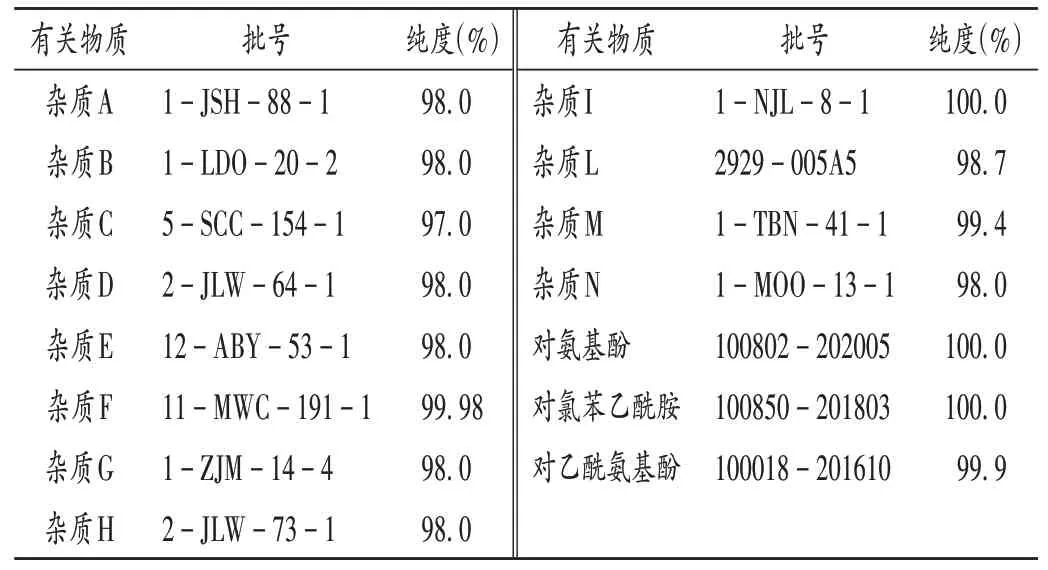
表2 有关物质对照品信息Tab.2 Information of references of related substances
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Xbridge C18柱(250 mm×4.6 mm,5µm);流动相:0.1%冰醋酸水溶液(A)-甲醇(B),梯度洗脱(程序见表3);流速:0.8 mL/min;检测波长:254 nm;柱温:30℃;进样量:5µL。在此色谱条件下的系统适用性试验结果见表4。

表3 流动相梯度洗脱程序(%)Tab.3 Program of gradient elution of mobile phase(%)

表4 系统适用性试验结果Tab.4 Results of the system suitability test
2.2 溶液制备
取甲醇150 mL,加水850 mL,混匀,作为稀释剂。
取对乙酰氨基酚片,研磨成粉,取细粉适量(相当于对乙酰氨基酚100 mg),精密称定,置10 mL容量瓶中,用稀释剂稀释并定容,振摇使溶解,滤过,取续滤液,即得供试品溶液。
取供试品溶液1.0 mL,置100 mL容量瓶中,用稀释剂稀释并定容;取1.0 mL,置20 mL容量瓶中,再次用稀释剂稀释并定容,摇匀,即得自身对照溶液。
取有关物质对氨基酚、对氯苯乙酰胺、杂质A-I和杂质L-N对照品各适量,精密称定,加稀释剂制成质量浓度为0.5 mg/mL的混合对照品贮备液。取混合对照品贮备液1.0 mL,置100 mL容量瓶中,加稀释剂稀释并定容,摇匀,即得混合对照品溶液。
取对氨基酚对照品5 mg,精密称定,置100 mL容量瓶中,用稀释剂稀释并定容;取1.0 mL,置100 mL容量瓶中,再次用稀释剂稀释并定容,摇匀,即得对氨基酚对照品溶液。
取对氯苯乙酰胺对照品溶液5 mg,精密称定,置250 mL容量瓶中,用稀释剂稀释并定容;取1.0 mL,置200 mL容量瓶中,再次用稀释剂稀释并定容,摇匀,即得对氯苯乙酰胺对照品溶液。
2.3 测定方法
精密量取2.2项下供试品溶液与各对照品溶液适量,分别注入高效液相色谱仪,按外标法以峰面积计算对氨基酚与对氯苯乙酰胺的含量,以加校正因子的自身对照法计算其他杂质的含量。
2.4 方法学考察
专属性试验:取2.2项下供试品溶液和混合对照品溶液各适量,置同一容量瓶中,混匀,按2.1项下色谱条件进样测定,记录色谱图(图1)。可见,对乙酰氨基酚色谱峰与各有关物质色谱峰均能达到基线分离,且分离度良好。

图1 专属性试验高效液相色谱图Fig.1 HPLC chromatograms of the specificity test
线性关系考察与相对校正因子:取对乙酰氨基酚和有关物质对照品各适量,精密称定,配制系列质量浓度的标准曲线溶液,按2.1项下色谱条件进样测定,以峰面积(Y)为纵坐标、各成分质量浓度(X,µg/mL)为横坐标进行线性回归,并按主成分线性方程的斜率比各杂质线性方程的斜率计算相对校正因子。结果见表5。
定量限与检测限确定:取2.2项下稀释剂,待仪器平衡后注入色谱仪,连续进样3次,记录基线噪音,根据系统适用性试验中各杂质的峰高逐级稀释,至信噪比(S/N)分别为10±1和3±1,得到各杂质的定量限和检测限。结果见表5。

表5 方法学考察结果与相对校正因子Tab.5 Results of the methodological investigation and RCFs
中间精密度试验:在同一实验室,由不同实验员在不同日期进行重复性试验。结果的RSD均小于4.0%,表明方法中间精密度良好。
稳定性试验:取2.2项下供试品溶液与混合对照品溶液,分别于室温下放置0,4,8,12,24 h时按2.1项下色谱条件进样测定,记录峰面积。结果供试品溶液与混合对照品溶液中各有关物质峰面积的RSD均小于3.0%(n=5),表明供试品溶液与对照品溶液在室温下放置24 h稳定性良好。
重复性试验:取对乙酰氨基酚片,研磨成粉,取细粉适量(相当于对乙酰氨基酚100 mg),精密称定,置10 mL容量瓶中,加入2.2项下混合对照品贮备液1.0 mL,用稀释剂稀释并定容,振摇,滤过,取续滤液,平行6份,按2.1项下色谱条件分别进样测定,按2.3项下方法计算。结果的RSD均小于2.0%(n=6),表明方法重复性良好。
加样回收试验:取对乙酰氨基酚片,研磨成粉,取细粉适量(相当于对乙酰氨基酚100 mg),精密称定,共9份,置10 mL容量瓶中,分别加入80%,100%,120%限度的杂质A-I、杂质L-N、对氨基酚、对氯苯乙酰胺对照品,各3份,用稀释剂稀释并定容,摇匀,滤过,取续滤液,按2.1项下色谱条件进样测定,记录峰面积,并计算加样回收率。结果见表5。
破坏试验:取2.2项下供试品溶液,在酸(1 mol/L盐酸溶液2 mL破坏2 h)、碱(1 mol/L氢氧化钠溶液2 mL破坏2 h)、高温(90℃水浴破坏2 h)、氧化(30%过氧化氢溶液2 mL破坏2 h)及光照(4 500 lx破坏2 h)条件下进行强制降解试验。结果对乙酰氨基酚在酸、碱、氧化及光照条件下,降解过程中均未产生新的杂质;高温条件下降解产生了新的杂质,各杂质峰均基线分离,表明方法专属性良好。色谱图见图2。

图2 破坏性试验高效液相色谱图1.Paracetamol 2.Impurity B 3.Unknown impurity 4.Impurity G 5.Impurity H a.Destroyed by high temperature b.No destroying c.Destroyed by acid d.Destroyed by alkali e.Destroyed by oxidation f.Destroyed by lightFig.2 HPLC chromatograms of the destructive test
耐用性试验:取2.2项下供试品溶液与混合对照品溶液各适量,分别考察不同批次色谱柱,不同柱温(25,30,35℃),不同流速(0.7,0.8,0.9 mL/min)和不同检测波长(250,254,258 nm)条件下各有关物质的分离度。结果各有关物质色谱峰的分离度均大于1.5,含量的RSD均小于4.0%,表明方法耐用性良好。
2.5 样品有关物质含量测定
取12批(编号为Ⅰ-Ⅻ)样品各适量,精密称定,按2.2项下方法制备供试品溶液,按2.1项下色谱条件进样测定,按2.3项下方法计算各样品中有关物质的含量。结果12批不同厂家的样品中14个有关物质含量为0.008 0%~0.070 4%。对乙酰氨基酚片参比制剂(编号为Ⅰ)中检出杂质B与杂质H;国内11批(编号为Ⅱ-Ⅻ)对乙酰氨基酚片中检出对氨基酚、杂质A-D、杂质F、杂质H、杂质L、杂质N,均未检出对氯苯乙酰胺、杂质E、杂质G、杂质I、杂质M。详见表6。
3 讨论
3.1 色谱条件选择
《欧洲药典》10.0版中关于对乙酰氨基酚有关物质的分析较全面,但以HALO-C18柱(100 mm×2.1 mm,2.7µm)为色谱柱时杂质A与杂质B未能完全分离。由于色谱柱内径及粒径较小,液相色谱仪的柱压较高,长期使用会对仪器造成不良影响,且色谱柱的使用寿命不佳。调整色谱柱为Xbridge-C18柱(250 mm×4.6 mm,5µm),流动相为纯化水-甲醇,但仅能分离13个杂质,且对氨基酚与对乙酰氨基酚的色谱峰峰形稍有拖尾,影响样品测定的准确性。最终将流动相调整为0.1%冰醋酸水溶液-甲醇,分离效果良好。
3.2 测定和计算方法选择
除对氨基酚与对氯苯乙酰胺外,国内外药典均采用自身对照法测定其他杂质的含量。但杂质A-F、杂质H-I、杂质L-M的相对校正因子分别为1.15,1.27,1.26,1.48,2.79,1.24,1.33,1.27,1.84,2.13,均不在0.9~1.1范围内,应采用更合理的带相对校正因子的自身对照法进行检测和计算,以保障杂质检测的准确性。
3.3 杂质限度确认
2020年版《中国药典》、《欧洲药典》10.0版和《美国药典》40版中均有收载对乙酰氨基酚有关物质的检测,其中《欧洲药典》10.0版中有关物质限度最严格。由表6可知,12家企业的对乙酰氨基酚片中的有关物质的含量均未超标。参照《欧洲药典》10.0版制订对乙酰氨基酚片有关物质限度,即对氨基酚不得过0.005%,对氯苯乙酰胺不得过0.001%,杂质A-I与杂质L-N均不得过0.05%,总杂质不得过0.2%。
3.4 样品中杂质来源分析
由于对乙酰氨基酚一般以对硝基苯酚为原料,还原为对氨基苯酚后经乙酰化制得,其中对硝基苯酚由氯苯经硝化反应制得。由表6可知,对乙酰氨基酚片中有关物质检出量大于0.01%的杂质主要为杂质A、杂质B、杂质C、杂质H。其中,杂质A为对硝基苯酚原料中邻硝基苯酚导致的杂质,杂质B为丙酰化杂质,杂质C由对硝基氯苯原料氯苯中的邻二氯苯引入,杂质H为对氨基苯酚过度乙酰化产生。样品(编号Ⅰ)为仿制药一致性评价的参比制剂,其杂质含量明显低于国内大部分制剂。可见,对乙酰氨基酚片中有关物质主要来源于原料药合成过程中引入的杂质,并非由存储环境或辅料导致,故应从源头对原料药的质量进行控制,以提升制剂的质量。
3.5 方法评价
本研究中建立了有效检测对乙酰氨基酚片中有关物质的高效液相色谱法,方法专属性强、灵敏度和准确度均高,可有效分离14种有关物质及主成分;并考察了市售对乙酰氨基酚片及仿制药一致性评价参比制剂的有关物质,可为相关生产厂家及检验机构提供参考,有助于提升国内对乙酰氨基酚片的质量。
