致幻剂激活5-HT2AR产生致幻作用的信号通路研究进展
2022-11-09朱慧丽周培岚傅风华苏瑞斌
朱慧丽,周培岚,傅风华,苏瑞斌
(1.烟台大学药学院,山东 烟台 264005;2.军事科学院军事医学研究院毒物药物研究所,抗毒药物与毒理学国家重点实验室,北京 100850)
致幻剂是一类天然或人工合成的精神活性物质,能够影响人的中枢神经系统,可以强烈改变人的知觉、情绪和认知,使人产生错觉、幻觉以及情感改变,甚至导致自我歪曲、妄想症发作和精神思维分裂[1]。根据化学结构,致幻剂广义上可以分为两大类:吲哚烷胺类[如赛洛西宾、麦角酰二乙胺(lysergic acid diethylamide,LSD)]和苯烷胺类[如麦司卡林、2,5-二甲氧基-4-甲基苯丙胺(2,5-dimethoxy-4-methylamphetamine,DOM)、2,5-二甲氧基-4-碘苯丙胺(2,5-dimethoxy-4-iodoamphet-amine,DOI)][2]。虽然致幻剂激活的受体众多,但是目前普遍认为致幻剂主要通过激活或部分激活5-HT2AR产生致幻作用[3-4]。一些非致幻化合物,如麦角乙脲(lisuride,LIS)和麦角胺(ergotamine,ERG)与强效致幻剂LSD结构非常相似,它们与5-HT2AR有较强的亲和力,并能进一步激活受体[4],但是这些药物或化合物在临床使用时并未引起幻觉,在啮齿类动物也未观察到致幻剂所引起的标志性反应——甩头反应(head twitch response,HTR);它们对小鼠的基因转录图谱、电生理、受体后信号转导与经典致幻剂有不同的影响。此外,美国加州大学戴维斯分校的研究者们对致幻剂进行结构改造,得到了同样可以激活5-HT2AR的非致幻类似物。并且此类似物对药物成瘾、抑郁症和其他精神疾病都具有较好的治疗作用[5]。为什么只有特定的5-HT2AR激动剂引起致幻作用?以及哪些信号通路介导了致幻作用?这些尚无定论。本文综述了致幻剂激活5-HT2AR后相关致幻通路的研究进展,为早日揭开致幻作用的神秘面纱提供参考。
1 致幻剂概述
致幻剂使用历史悠久,在许多早期文明中,自然界中的致幻物质对哲学、宗教思想的形成和发展起着重要的作用[1]。致幻剂因能使人的意识发生深刻地变化,可作为神经、精神科学研究的工具。在科研和医疗领域,也表现出巨大的应用潜力,最初作为一种实验工具(拟精神病药物),用于制造精神分裂症模型;也曾作为抑郁症、创伤后应激障碍和物质使用障碍等精神疾病的治疗药物;在自身免疫性疾病、抗炎、镇痛、阿尔茨海默病、厌食症等方面也具有积极的作用[6]。目前,致幻剂辅助心理疗法显示出巨大的医疗前景。令研究者们振奋的是,与传统抗抑郁药物(单胺氧化酶抑制剂、三环类抗抑郁药、选择性五羟色胺再摄取抑剂等)相比,致幻剂能产生很强的神经可塑性这可能是它快速且持久的抗抑郁作用机制[7]。致幻剂一次给药治疗效果可持续十数天甚至数十天以上,进一步提高了患者的用药依从性和安全性[8]。不足之处是,在临床试验中,致幻剂因其强大的心理效应,需要在特定的医疗场所和专业的心理治疗师的支持和监督下进行,严重制约了其应用和发展。
近年来致幻剂作为新型毒品以不同的形式如“邮票”(主要成分是LSD)频繁出现在社会中,使我国一些认识不足的青少年深受蒙骗,纷纷涉足。目前,由于知识传播的迅速、材料的易得和法律的滞后,新型毒品的管理难度进一步加大,不法分子对违禁药物的分子结构稍微做一些改变或修饰,就可以产生效果相当或者作用更强的新毒品,以便于逃避法律的监管[9]。对毒品进行管理和司法打击常常呈现“道高一尺,魔高一丈”的较量,迫切需要合适的方法对药品和毒品进行判定。因此了解致幻剂的致幻机制显得至关重要,为趋利避害更好的运用此类化合物提供理论依据。
目前致幻剂的研究方法较为局限。现阶段幻觉还无法用生物学指标进行量化,也无法用标准化的语言进行描述。研究致幻剂对人体的致幻效应最常用的方法就是进行调查问卷,如《心理状态问卷》、《致幻剂评定表》、《意识状态变化评定表》等[10]。随着技术的进步一些成像方法也运用其中,例如正电子发射型计算机断层显像、功能磁共振成像。致幻剂对人的影响复杂、多变,很难确定具体的致幻动物模型;实验动物无法像人类那样填写调查问卷,也不可能用语言去描述,只能依靠动物的一些行为来预测人类特定的神经心理效应。目前,用于临床前研究的实验动物模型主要有药物辨别、HTR、前脉冲抑制、行为模式实验等[1]。人体和实验动物均证实致幻剂发挥致幻作用的主要靶点为5-HT2AR。对受体下游信号通路的进一步研究将有助于寻找新的靶标或生物标志物。对致幻作用的治疗和检测都有重要意义。
2 致幻剂的致幻靶点—5-HT2AR
5-HT2AR属于A型(视紫红质样受体)家族的G蛋白偶连受体(G protein-coupled receptors,GPCRs)。最初5-HT2AR因可介导各种类型平滑肌的收缩,不可逆拮抗剂“Dibenzyline”能阻断这一作用,Gaddum和Picarelli称其为D受体,后来Peroutka和Snyder根据与特定放射性配体的亲和力大小将其归为5-HT2受体。目前根据遗传同源性和一级结构特点,5-HT2受体包括5-HT2AR与5-HT2BR、5-HT2CR三个亚型,同源性达50%。5-HT2AR基因位于人类染色体13q14-q21上,人类、大鼠、小鼠的5-HT2AR包含471个氨基酸[11]。
5-HT2AR在外周和中枢神经系统广泛分布,在外周主要分布于胃肠道、心血管系统等。在中枢神经系统主要分布于新皮质、内嗅皮质、杏仁核、屏状核以及下丘脑;在海马、尾状核、壳核、伏隔核次之;而在丘脑、脑干、脊髓和小脑分布较少。5-HT2AR主要富集于大脑前额叶皮质第V层椎体神经元的顶端树突,目前认为该区域的5-HT2AR与致幻密切相关[11]。5-HT2AR的内源性配体为5-羟色胺(5-hydroxytryptamine,5-HT),对各种生理功能起着重要作用。5-HT对认知、感觉加工等的影响与激活中枢神经系统5-HT2AR密切相关。许多抗抑郁药、抗焦虑药和非典型抗精神病药物等通过作用于5-HT2AR而发挥作用。5-HT2AR的激活除产生幻觉外,对病理状态下中枢系统的调节也是近年来人们关注的重点。
3 致幻剂激活5-HT2AR后的信号通路
过去20多年来,人们越来越认识到,GPCRs除存在经典的激活“(R*)”和失活“(R)态外,一些化合物可以稳定不同的受体构象,从而优先激活特定的信号转导通路,这种现象被称为功能选择性或偏向性。5-HT2AR是偏向性研究最早的受体之一[11]。近期发表在《细胞》杂志上的一项研究成果更进一步肯定了此认识。加州大学戴维斯分校的研究人员开发出一种名为“PsychLight”的新型荧光传感器,这种基因编码的荧光传感器可以预测结构相似的5-HT2AR配体的致幻行为,当PsychLight与5-HT或致幻配体结合时,会稳定其特定构象,导致荧光增强。非致幻配体也可以与PsychLight结合,但会形成不同的荧光图谱[12]。近年来国内外对5-HT2AR后信号通路进行了大量的研究,发现其可与多种G蛋白和β-arrestins结合,如Gαq/11、Gαi/o和Gα12/13、β-arrestin2;从而进行不同途径的信号转导。虽然这些激活的信号通路的分子细节和药理作用还不清楚,但对进一步阐明致幻作用的信号通路具有重要的提示作用。
3.1 Gαq/11-PLCβ信号通路最典型的5-HT2AR后信号通路为Gαq/11-PLCβ信号通路。5-HT2AR与Gαq/11偶联,使磷酸肌醇(phosphoinositide,PI)特异性磷脂酶Cβ(phospholipase Cβ,PLCβ)活化,在PLCβ的催化作用下促使细胞膜上的磷脂酰肌醇-4,5-二磷酸在sn-3位置水解,产生肌醇-1,4,5-三磷酸(inositol-1,4,5-triphosphate,IP3)和二酰基甘油(diacylglycerol,DAG)。IP3使细胞内钙库的Ca2+释放,激活钙/钙调蛋白依赖性激酶II(calcium/calmodulin dependent kinase II,CaMKII)。DAG与膜结合并激活蛋白激酶C(protein kinase C,PKC),PKC途径可磷酸化胞外信号调节激酶1/2(extracellular signal-regulated kinase,ERK)和转录因子环腺苷酸反应元件结合蛋白(the transcription factor cyclic AMP response element-binding protein,CREB)等,以调节基因表达[11,13]。
长期以来,人们认为Gαq-PLCβ-PI级联反应与致幻作用最相关,但这一假设存在诸多矛盾。在常用于评价致幻剂的HTR模型上,基因敲除5-HT2AR或者使用5-HT2AR拮抗剂均可阻断致幻剂全身给药诱导的小鼠HTR;使用遗传策略恢复小鼠皮质锥体神经元上5-HT2AR的表达,可以挽救致幻剂诱导的HTR[4]。5-HT2AR激动剂局部注射到内侧前额叶皮质可诱导大鼠产生HTR。因此,5-HT2AR在啮齿类动物大脑前额叶皮质中的激活是致幻剂诱导HTR的必要条件。但5-HT2AR下游信号分子Gαq的基因敲除后,与野生型(wild-type,WT)小鼠相比,DOI诱导的HTR总次数仅降低了35%~40%[14]。Gα11蛋白与Gαq在结构和功能上非常相似,在细胞内信号级联反应中可相互替代。由于Gα11在大脑各个区域的表达量均明显低于Gαq。即使存在Gα11的代偿,Gαq基因敲除后DOI诱导的HTR降低不到50%,不排除其他信号通路参与了DOI诱导的HTR。非致幻剂LIS激活5-HT2AR后也能与Gαq偶联,Banerjee等[13]发现DOI激活Gαq信号通路的程度较LIS明显增强,在表达人/大鼠5-HT2AR-EGFP的HEK293细胞和新生Sprague-Dawley大鼠原代皮层神经元中,与非致幻剂LIS相比,DOI剂量依赖性的增加PLCβ、PKC、ERK、CaMKII、CREB的磷酸化,并且胞内IP3和DAG的水平也较高。以上文献资料提示Gαq信号通路并不是致幻剂激活5-HT2AR产生致幻效应的特异性通路,但仍然是致幻效应完整显现所必需的信号通路。
PLCβ为Gαq/11与5-HT2AR解偶联后的第一激活酶,其抑制剂U73122可完全阻断LSD和LIS诱导的即刻早期基因c-fos,U73122也能抑制LSD诱导的早期生长反应因子1(early growth response factor 1,egr-1)和早期生长反应因子2(early growth response factor 2,egr-2)的转录[4]。进一步说明经典的Gαq/11-PLCβ信号通路不是致幻剂特异性激活信号通路。
5-HT2AR激活后PI水解与幻觉产生的相关性也存在疑问。在人类和啮齿类动物中,LSD类致幻剂与5-HT2AR的亲和力与它们的致幻效能有很强的相关性。但是,Roth等[15]发现LSD类致幻剂与5-HT2AR亲和力的高低与受体后激活PI水解的效能之间没有明显的相关性。并且在稳定表达大鼠5-HT2AR的PC12细胞,LSD与5-HT、DOM相比,在激活PI水解方面虽然效价强度高,但效能较低[16]。此外,Rabin等[16]注意到,在药物辨别试验中,一些致幻剂在药物替代中的效能与它们激活PI水解的效能之间缺乏相关性。他们认为PI水解可能不是参与药物辨别试验中替代效应的关键信号通路;致幻剂激活5-HT2AR产生超过阈值水平的PI水解可能只是引起致幻作用的几个必要因素之一,并不是唯一的关键因素[16]。
一般认为,Gαq/11激活PLCβ后水解PIP2产生IP3,引起胞内Ca2+动员。在新生大鼠皮质原代培养的Ⅰ型星形胶质细胞,5-HT2AR拮抗剂凯坦色林或4-(4-氟苯甲酰基)-1-(4-苯基丁基)哌啶草酸酯能够抑制5-HT诱发的[Ca2+]i升高[17]。百日咳毒素(pertussis toxin,PTX)预处理后,对5-HT引起的Ca2+动员没有明显影响,说明Gαi、Gαo、Gαt不介导此通路。PLC抑制剂U73122和新霉素可阻断5-HT诱导的[Ca2+]i升高[17-18],IP3受体拮抗剂肝素单独给药可抑制5-HT诱导的[Ca2+]i升高[17]。此外,有研究结果表明Gαq信号途径可能不是Ca2+动员的唯一决定因素。Cussac等人[19]在稳定表达人5-HT2AR的CHO细胞,通过[35S]GTP-γS检测5-HT2AR激动剂对Gαq/11蛋白的激活,应用荧光探针(Fluo-3)检测胞内[Ca2+]i的变化,比较激动剂的EC50值,发现内源性配体5-HT和5-羧基色胺(5-carboxytryptamine,5-CT)作用后诱导的Ca2+释放强于对Gαq/11蛋白激活,效价强度前者为后者的20至50倍;1-(3-氯苯基)哌嗪[1-(3-chlorophenyl)piperazine,mCPP]和DOI对Ca2+释放与Gαq/11蛋白激活的效价强度相当,前者为后者的2至3倍;而LSD激活Gαq/11蛋白比诱导Ca2+释放的效价强度高20倍;非致幻剂LIS对两种途径均表现出部分激动剂的特性,对Gαq/11激活的选择性更高,是Ca2+释放的1 000倍以上。(见Tab 1)
在新生大鼠的皮质星形胶质细胞,加入5-HT(10 μmol·L-1)后引起细胞内钙库的Ca2+大量释放,与5-HT2AR下游IP3激活密切相关,[Ca2+]i升高进一步引起胞外钙离子内流[17-18]。但Ca2+动员引发的细胞外Ca2+内流的机制目前存在一定的分歧,Jalonen等[18]认为钙库释放后进一步激活了胞膜上的蜂毒敏感的小电导Ca2+激活K+通道,随后L型Ca2+通道被激活(仅当细胞外液有Ca2+存在时被激活,可被L型Ca2+通道阻滞剂尼莫地平和硝苯地平抑制),引起了外钙内流。Hagberg等[17]则认为,5-HT引起的外钙内流不是L、N或T型电压调控钙通道开放的结果,而是与耗竭型Ca2+通道有关。5-HT、DOM、DOI、LSD等激活5-HT2AR后剂量依赖性的增加细胞内[Ca2+]i水平,目前胞内[Ca2+]i的增加和胞外Ca2+的内流都是在离体状态下检测的结果。尽管不同激动剂激活Gαq/11和升高[Ca2+]i的效能与其致幻效能之间没有相关性[19],但Cussac等[19]认为[Ca2+]i水平的上调对致幻阈值可能很重要。
3.2 Gαi/o介导的信号通路配体作用于5-HT2AR后,还可能激活PTX敏感的Gαi/o信号通路。研究表明,致幻剂和非致幻剂在此通路上有诸多差异。大多数信号通路最终通过调控基因表达以响应细胞外刺激。致幻剂LSD和非致幻剂LIS对小鼠中枢神经系统c-fos、egr-1和egr-2等基因转录的影响不同。除海马外,在不同大脑区域嗅球、前额叶皮质、扣带回皮质、躯体感觉皮质、梨状皮质、海马、丘脑和小脑,LSD和LIS都能激活c-fos。LSD能够增加皮质和嗅球egr-1和egr-2的转录水平,而在丘脑和小脑等脑区没有作用。LIS对所有脑区egr-1和egr-2的转录水平都没有影响。PTX预处理可显著降低LSD诱导的egr-1和egr-2增加。这提示egr-1和egr-2的基因应答具有致幻剂作用特异性,并且这一变化与Gαi/o信号传导有关[4]。在HEK293细胞中,定量磷蛋白组学发现致幻和非致幻剂激动5-HT2AR后诱导不同模式的蛋白磷酸化。PTX预处理可抑制致幻剂DOI和LSD诱导的ERK1/2磷酸化,而不影响非致幻剂LIS和ERG诱导的反应[20]。结合致幻剂和非致幻剂对基因转录水平的影响,提示Gαi/o偶联的信号通路可能是区分致幻与非致幻的重要通路。
5-HT2AR激活后,不仅引起Gαq-PLCβ-PI级联反应,还可激活磷脂酶A2(phospholipase A2,PLA2),该酶优先水解在sn-2位上含有磷脂的花生四烯酸,从而生成游离花生四烯酸(arachidonic acid,AA)和溶血磷脂。多个细胞系和小鼠胚胎的海马切片证实PLA2途径独立于PLC信号转导途径[21-22]。进一步研究发现PLA2信号转导途径比PI转换级联更为复杂,在NIH3T3细胞中涉及多个G蛋白,Kurrasch-Orbaugh等对PLA2-AA信号通路进行了详细的探究,他们发现Gαi/o信号通路和Gα12/13信号通路一起共同介导了AA释放。Gαi/o信号通路激活后,Gαi/o和Gβγ三聚体解离,Gβγ独自启动Ras-Raf-MEK-ERK信号级联,ERK1/2磷酸化后激活PLA2,生成游离AA。Gα12/13信号通路激活后,主要通过Rho信号级联反应激活p38 MAPK,p38 MAPK进一步激活PLA2[23]。但是,Gα12/13信号通路与致幻相关研究较少。
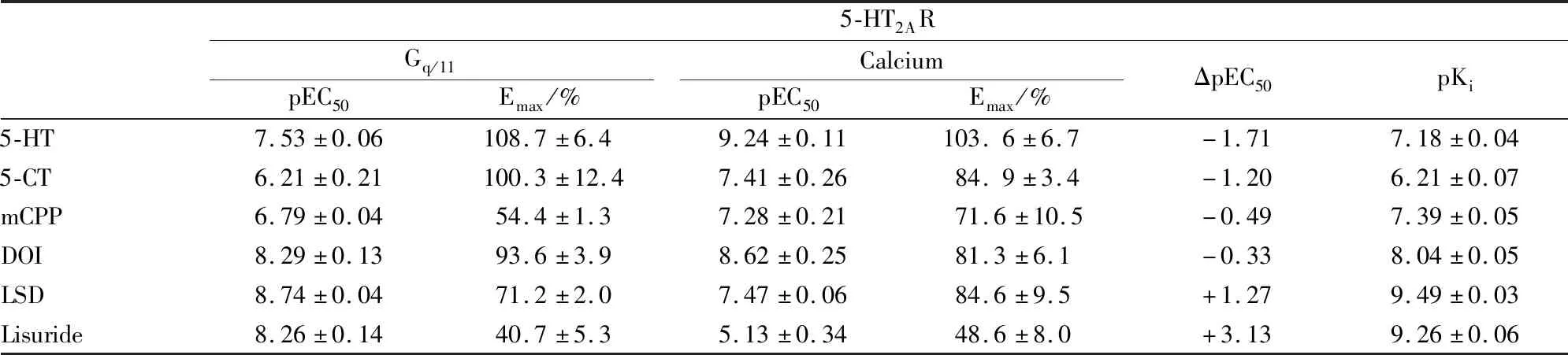
Tab 1 Stimulation of [35S] GTPγS binding to Gq/11 proteins and stimulation of Ca2+ release by agonists at h5-HT2A receptor[19]
尽管PLA2这一通路的意义尚未被详细研究,但Qu等人[24]通过定量放射自显影来测量静脉注射放射性标记的花生四烯酸酯([3H]AA)的脑掺入,对PLA2的激活进行成像观察。发现给大鼠2.5 mg·kg-1DOI可显著增加神经元[3H]AA的纳入,尤其是在新皮层[3H]标记的AA掺入大量增加,并且5-HT2AR密度越高的区域增加越多。不同激动剂作用于5-HT2AR后,激活PLC和PLA2通路的程度有所不同。Berg团队首次证明[21],在CHO细胞表达5-HT2AR后,不同激动剂对具体信号转导途径激活的相对效率有所不同:与5-HT相比,激动剂3-三氟甲基苯基哌嗪优先激活PLC-IP途径,而LSD则显示出对PLA2-AA途径的偏好。在NIH3T3细胞中表达大鼠5-HT2AR后,LSD、DOB、二甲-4-羟色胺和5-MeO-DMT激活AA和PI水解通路的EC50值不同[22]。在这项研究中,大部分致幻类化合物诱导AA释放的效价强度都比刺激PI水解的高。例如,在二甲-4-羟色胺激活AA释放的效价强度是刺激PI水解的近30倍。这些研究结果提示,花生四烯酸的产生与致幻作用可能有更明显的相关性。
3.3 β-arrestin介导的信号通路β-arrestins是一种细胞内支架蛋白,可以抑制或促进GPCR信号的传递。β-arrestins在GPCR被G蛋白偶联受体激酶磷酸化后募集至受体上,作为脚手架蛋白触发网格蛋白介导受体内化。β-arrestins还可能通过阻止受体与G蛋白偶联从而抑制信号转导,成为GPCR的负调控因子。对于某些GPCR,β-arrestin还以G蛋白非依赖的方式激活MAPK、Src蛋白酪氨酸激酶、磷脂酰肌醇3激酶(phosphoinositide 3-kinase,PI3-K)和蛋白激酶B(Akt)等多种信号转导途径。
在体外,β-arrestin1和β-arrestin2都能与编码大鼠5-HT2AR第三胞内环的融合蛋白相互作用。在HEK293细胞,共转染小鼠5-HT2AR(HA-5-HT2AR)和β-arrestin2(βarr2-GFP)后,5-HT能够诱导β-arrestin2向质膜易位。同样在HEK293细胞,给予5-HT后,发现二者存在相互作用。在体内,β-arrestin1和β-arrestin2与5-HT2AR共定位于大鼠前额叶皮质锥体神经元,在一些细胞内囊泡中β-arrestin1和5-HT2AR也存在一些共定位。5-HT的前体5-羟色氨酸(5-hydroxytryptophan,5-HTP)作用于小鼠后,额叶皮质的5-HT2AR与β-arrestin2也存在相互作用[25]。但是,β-arrestins与5-HT2AR的作用因激动剂而异,在小鼠胚胎成纤维细胞,5-HT诱导5-HT2AR的内化是β-arrestin依赖性的,而DOI诱导的5-HT2AR内化是非β-arrestin依赖性的[26]。
在评估激动剂导向的信号通路时,不同的激动剂β-arrestins的作用也不同。在小鼠胚胎成纤维细胞,5-HT可以通过β-arrestin和Gαq-PLC两条途径促进ERK1/2磷酸化,而DOI只通过Gαq-PLC途径促进ERK1/2磷酸化[26]。这种差异在体内也进行了评估,5-HTP和DOI作用于WT小鼠后,都可诱导额叶皮质ERK1/2磷酸化;在β-arrestin2敲除后,5-HTP诱导的ERK1/2磷酸化程度大大降低,DOI诱导的磷酸化不受影响,这与体外的研究结果一致。表明内源性配体5-HT激活5-HT2AR引起ERK1/2磷酸化是β-arrestin2依赖性的,而DOI是非依赖性的[26]。这些差异也反映在动物行为上,给予5-HT的前体5-HTP,可诱导WT小鼠产生HTR,在β-arrestin2敲除小鼠上则不引起HTR,DOI无论在WT还是β-arrestin2敲除小鼠都引起HTR。表明5-HT引起的HTR是β-arrestin2信号通路依赖型的,而DOI诱导的HTR是β-arrestin2非依赖性的。进一步研究发现,5-HT可能通过激活β-arrestin2-PI3-K-Src-Akt信号通路产生HTR[25],DOI则不激活此通路。此外,同一实验室的研究者们还发现,非典型抗精神病药物氯氮平作用于小鼠后,通过激活5-HT2AR下游β-arrestin2非依赖的Akt途径而诱导抗精神病样的行为反应[27]。
以上结果提示,5-HT2AR与β-arrestin2的相互作用对响应内源性5-HT功能起重要作用,下游的Akt在5-HT、DOI和氯氮平三类化合物中所起的作用各异,对致幻机制的探究具有重要的指导意义,需要进一步阐明其具体作用。
3.4 PLD信号通路NpxxY是一种特殊的保守序列,存在于包括5-HT2AR在内的许多视紫红质样GPCRs的TM7和羧基端结构域之间,被认为是ADP-核糖基化因子(ADP-ribosylation factor,ARF)介导的信号决定簇。5-HT2AR可以通过独立于Gαq/11信号通路、ARF依赖的方式激活磷脂酶D(Phospholipase D,PLD)。免疫共沉淀试验和ARF阴性突变都表明,ARF1在PLD通路中发挥关键作用。进一步研究表明,TM7中的N376PxxY结构域对ARF依赖的PLD信号转导和与ARF1的相互作用是必需的。此外,ARF1通过GTP依赖性与5-HT2AR的羧基端相互作用,参与了这一机制[11,28]。
4 结语与展望
尽管致幻剂激活的受体众多,现在越来越多的数据支持致幻剂的致幻作用是由5-HT2AR介导的,可能有其他受体也参与其中,从而形成了复杂的调控网络。据目前的研究结果来看,5-HT2AR后信号通路的复杂程度一点也不亚于致幻剂激活的受体。除了较为经典的Gαq/11、Gαi/o和Gα12/13、β-arrestin2通路,还有众多蛋白及蛋白激酶参与调控[11]。目前评价幻觉的动物模型有限,大量的研究仅依靠分子水平的差异去寻找致幻信号通路,是远远不够的。再加上检测指标的缺乏更加大了致幻剂研究的难度。
为了更好的确定5-HT2AR激活后的致幻信号通路,在其他致幻模型上还需要进一步的验证。体内和体外实验相结合,致幻剂与非致幻剂相对照,对5-HT2AR下游信号通路的认识会越来越明朗,尤其是动物行为学的支持。目前的研究进展也提供了重要的数据基础和理论依据。对致幻机制的阐明、药物靶标的发现和生物标志物的确立具有重要的指导意义。
