肺动脉高压致病基因和靶向治疗研究进展
2022-11-07王蔚钟子晴荀秋芬祝国风杨青
王蔚,钟子晴,荀秋芬,祝国风,杨青
肺动脉高压(pulmonary hypertension,PH)是一组以毛细血管前PH为特点的疾病,其致残率和病死率均较高。从家族性PH基因研究到高通量组学技术,越来越多的致病基因和突变基因被发现,使研究者对PAH突变基因及发病机制的了解更加深入,也为寻找新的药物靶点提供了科学依据。本文主要综述了PAH致病基因和靶向治疗研究进展,以期为PAH的分子机制探索和个体化治疗提供思路。
1 PAH的分类和致病基因
在临床上,PAH可分为7个亚型,包括特发性肺动脉高压(idiopathic pulmonary hypertension,IPH)、遗传性肺动脉高压(heritable pulmonary hypertension,HPH)、药物和毒物相关性PH、疾病相关的PH、对钙通道阻滞剂长期有效的PH、具有明显肺静脉/肺毛细血管受累的PH〔遗传性肺毛细血管瘤病(pulmonary capillary hemangiomatosis,PCH)和肺静脉闭塞病(pulmonary veno-occlusive disease,PVOD)〕和新生儿持续性PH[1]。目前,通过测序技术发现了16个PH的致病基因[2-19](见表1),这些致病基因功能异常可引起不同的PH表型。
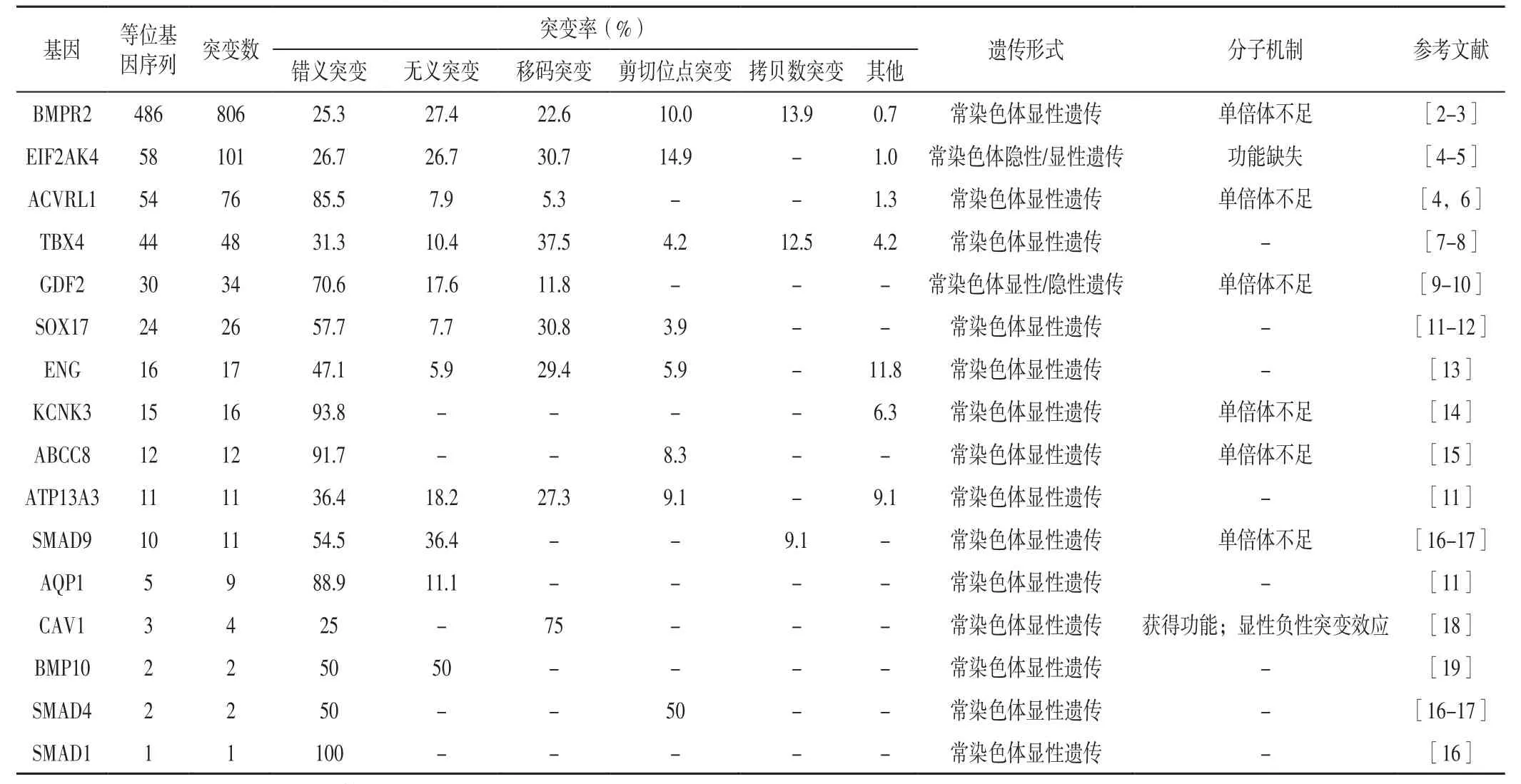
表1 PH的致病基因Table 1 Pathogenic genes of PH
2 与IPH和HPH相关的突变基因
2.1 转化生长因子β(transforming growth factor-β,TGF-β)信号通路致病基因
2.1.1 骨形成蛋白受体2(bone morphogenetic protein receptor type 2,BMPR2) 2000年,研究者发现了PH的首个致病基因——BMPR2,其是TGF-β超家族成员,是PH(特别是IPH和HPH)的主要发病基因[20]。
2.1.1.1 BMPR2的结构及突变类型 BMPR2可编码TGF-β家族Ⅱ型受体,在肺动脉内皮细胞(pulmonary arterial endothelial cells,PAECs)上呈高表达。目前,在MalaCards数据库(https://www.malacards.org/),研究者从800多例PH患者中检测到486个不同位点的非重复性突变基因,这些突变位点广泛分布于外显子中,且大多数位于关键功能区,其中25.3%是由氨基酸替换产生的错义突变、27.4%是无义突变、22.6%是小核苷酸插入或缺失引起的移码突变、13.9%是拷贝数突变、10.0%是剪切位点突变[2-3,21]。
2.1.1.2 BMPR2功能 BMPR2蛋白可通过TGF-β信号通路对疾病产生作用,而TGF-β信号通路主要有两条传导路径:(1)TGF-β在细胞膜上与其受体结合形成复合物,诱导Ⅱ型受体磷酸化Ⅰ型受体ALK4、ALK5、ALK7蛋白,进而活化下游Smad2、Smad3蛋白;(2)骨形成蛋白(bone morphogenetic protein,BMP)与其Ⅱ型受体BMRP2等结合后,Ⅱ型受体可磷酸化Ⅰ型受体ALK1、ALK2、ALK3、ALK6蛋白,进而激活下游Smad1、Smad5、Smad8蛋白。Smad蛋白是一种转录因子,在信号传导过程中其一旦被磷酸化,则Smad2、Smad3或Smad1、Smad5、Smad8会与Smad4形成复合物并转移到细胞核中,进而调节下游靶基因表达[22]。TGF-β和BMPR2受体基因突变破坏了TGF-β信号传导通路,最终导致TGF-β信号增强、BMPR2信号减弱,肺动脉增殖。
2.1.1.3 BMPR2突变的临床表型 BMPR2突变导致了70%~80%的HPH和10%~40%的IPH,患者主要表现为心脏指数偏低、肺血管阻力升高、平均肺动脉压升高及静脉血氧饱和度降低,患者对急性肺血管扩张试验的反应性差,疾病诊断及死亡的年龄更小;其中男性PH患者BMPR2突变率、死亡风险更高[23],而BMPR2突变的女性PH患者较无BMPR2突变的女性PH患者疾病诊断年龄更小,且错义突变患者较截断突变患者病情更严重[24]。
BMPR2突变的平均外显率仅为20%左右,且常见的p.S775N突变与PH发病无明显相关性,提示一些携带突变基因的个体并不会发病[10]。目前研究认为,环境及修饰基因对疾病外显可能具有重要作用,PH患者淋巴细胞中野生型BMPR2表达水平明显低于未发病的BMPR2突变携带者,进而影响BMP通路,故野生型BMPR2作为修饰因素可能影响致病基因的外显[25]。
除了基因外显率低之外,“二次打击”理论(即带有附加修饰基因的致病基因)被认为参与了PH的发病。1例携带BMPR2突变的PH患者,约20%的PAECs有13号染色体缺失,这段缺失的染色体包含BMP通路的下游基因Smad9,进而造成其编码的Smad8蛋白表达缺失,反过来Smad8蛋白表达缺失又使BMP信号通路功能进一步受损,即“二次打击”[26]。
2.1.2 其他致病基因 除BMPR2外,TGF-β信号通路对肺动脉平滑肌细胞增殖、细胞外基质合成和分化也具有重要作用[27]。
2.1.2.1 Smad家族 2011年,有研究者对324例散发性PH患者进行基因测序,发现Smad家族的Smad1、Smad4、Smad9发生了由氨基酸替换产生的突变[16],其中有两种错义突变在体外表现出信号传导活性降低。Smad4中一个剪切位点突变导致了因剪切效率降低而产生的转录丢失。DRAKE等[17]研究发现,Smad9杂合性缺失可明显影响Smad的转录活性。
2.1.2.2 BMP9突变 BMP9编码的生长分化因子2(growth differentiation factor 2,GDF2)是PAECs BMPR2/ACVRL1受体复合物的主要循环配体,而该受体信号传导对内皮分化和血管形态发生至关重要。有研究者对一组欧洲PH家系进行全基因组测序发现,BMP9基因杂合突变包括1个移码突变和7个错义突变[11];对一个中东PH家系进行全基因组测序发现了BMP9的新双等位基因突变(p.P85L和p.D218N),其与BMPR2(p.S775N)联合突变产生了该家族唯一发病表型[10];该测序结果同时验证了BMPR2基因的“二次打击”理论。一项采用基因面板的前瞻性研究发现,1.2%的成年PH患者存在BMP9突变[28]。一项针对中国331例PH患者全外显子组测序研究发现,6.7%的患者伴有BMP9突变[29]。上述研究初步表明,BMP功能缺失突变在PH发病中具有一定作用。
2.1.3 ACVRL1和ENG 对遗传性出血性毛细血管扩张症(hereditary hemorrhagic telangiectasia,HHT)家系合并PH患者进行基因组测序发现了ACVRL1和ENG致病突变,且绝大多数ACVRL1突变属于错义突变,其中约89%的突变位点位于重要的催化结构域[30]。ACVRL1突变后机体ALK1蛋白表达减少,进而影响功能激酶结构域,抑制PAECs中的Smad1、Smad5、Smad8通路活性,同时刺激ALK5活性及增加下游Smad2、Smad3信号通路,导致BMPR2信号减弱、TGF-β信号增强,最终促进细胞增殖和迁移。研究表明,ACVRL1突变携带者较无ACVRL1突变携带者和BMPR2突变携带者PH发病年龄更小[6],且部分PH患者诊断早于HHT的临床表现。虽然ACVRL1突变携带者在诊断时未出现明显的血流动力学改变,但其对急性血管扩张试验无反应,且疾病进展快、预后更差[31]。
ENG主要在PAECs上表达,其可作为共受体来稳定配体-受体复合物,并保证受体信号的正确传导。有研究者对一组ENG突变的PH患者进行基因组测序,发现16例患者有8个致病突变基因,最常见的是p.G191D突变;其中5例患者同时携带BMPR2、ACVRL1突变[13]。研究表明,ENG剪切位点突变导致单倍体功能不足,使受体信号传导障碍、血管生成失调、血管畸形及PAECs异常增殖[32];临床上ENG突变携带者平均PH诊断年龄较无ENG突变者小8岁[13]。
2.2 钾通道相关致病基因
2.2.1 KCNA5 KCNA5编码的Kv1.5是电压门控钾通道成员之一,其支持心房特异性钾电流。PH患者存在KCNA5突变,KCNA5和BMPR2双基因突变可引起肺动脉收缩和严重的早发PH表型。
2.2.2 KCNK3 KCNK3是编码钾通道蛋白超家族成员之一。2013年,研究者通过测序发现KCNK3的6种杂合子错义突变(G203D、G97R、V221L、T8K、E182K和Y192C),且均为致病突变[33]。2017年,有研究者在日本PH患者中发现了杂合子错义突变(p.G203D)[34]、在西班牙PHA患者中发现了纯合子突变(p.G106R和p.L214R)[35]。上述研究证实了KCNK3在PHA发病中的作用,KCNK3功能和表达缺失是PH患者右心室肥大和功能障碍的标志。研究表明,携带KCNK3纯合子突变的PH患者,发病年龄更小、病情更严重;但KCNK3突变为不完全显性遗传,且纯合子突变患者临床症状更严重[35]。
2.2.3 ABCC8 ABCC8可编码ATP敏感性钾通道调节亚基蛋白。对未发现BMPR2和ACVRL1突变的儿童和成年PH患者进行筛查,发现了ABCC8的一个新的有害错义突变(p.R958H),该突变位于编码蛋白的一个关键结构域的高度保守区,且ABCC8突变分别与IPH、HPH或冠心病-PH患者相关,提示该基因在PH各亚型中无特异性分布[15]。
3 与PCH和PVOD相关的致病基因
2014年,研究者在遗传性和散发性PCH[36]、PVOD[37]患者中均发现了EIF2AK4致病突变,其是编码磷酸化真核生物翻译起始因子2的α亚单位激酶。PCH和PVOD是PH的少见类型,均为常染色体隐性遗传。目前指南推荐,检测出EIF2AK4双等位基因突变就可以对遗传性PVOD/PCH进行诊断,不需要进行肺活检[38]。有研究者对1个HPH家系进行二代测序发现,家族成员均携带两种致病杂合子突变:BMPR2移码突变和EIF2AK4剪切位点新突变,且同时发生两种基因突变的家族成员才发病[39]。上述研究结果不仅支持“二次打击”理论,还提示对已知携带基因突变的家族成员,有必要进行PH的基因测序。
研究表明,EIF2AK4突变患者比无EIF2AK4突变患者的诊断年龄小、生存率低[5]。分析EIF2AK4突变患者治疗困难的原因是其对现有前列环素衍生物、内皮素受体拮抗剂和磷酸二酯酶5型抑制剂无良好的治疗反应,且易出现肺水肿。但药物治疗也存在有效案例,如1例散发性EIF2AK4突变的PVOD患者,经前列环素类似物治疗后其急性加重次数减少、体力改善、肺动脉高压得到控制[40]。此外,在IPH或HPH患者中,携带EIF2AK4双等位基因突变者平均发病年龄<50岁、一氧化碳弥散量<50%[4],提示携带EIF2AK4双等位基因突变的PH患者发病年龄小、肺弥散功能差。
4 与儿童PH相关的突变基因
研究表明,在儿童PH中发现的突变基因包括BMPR2、ACVRL1、ABCA3、NOTCH3、KCNK3、HTR2B、ENG和EIF2AK4,且基因突变携带者肺血管阻力指数较高,对血管扩张剂反应很差,1、2、3年生存率分别为86.6%、63.8%、52.2%[41]。近年来通过测序发现了更多与儿童PH相关的致病基因。
4.1 CAV1 CAV1可编码小窝蛋白1,在内皮细胞中呈高表达,是形成胞膜穴状内陷的结构蛋白。CAV1对BMP受体的物理结构起作用,能调节PAECs的功能和渗透性,其表达缺失会减弱BMPR2的膜定位和信号传导[42]。在PH患者肺动脉组织中,CAV1蛋白表达降低反映了肺血管内皮细胞增殖和抗凋亡表型。
4.2 TBX4 TBX4缺失和突变与小髌骨综合征相关[7],通常可导致氨基酸替换或转录过程过早截断。TBX4杂合子突变功能缺失是幼年早发PH最常见的遗传因素,可伴或不伴小髌骨综合征[43]。
5 其他致病基因
2018年,有研究者对先天性心脏病相关PH患者进行基因组测序,发现了SOX17 p.Y137基因突变引起编码蛋白截短的现象,提示SOX17是一个新的候选易感基因[44]。SOX17基因高度保守,是在发育过程中参与Wnt/β-catenin和Notch信号通路的转录因子,可调节细胞类型和组织分化,与β-catenin结合后可激活靶基因转录。多数SOX17突变是通过使结合结构域缺失或阻止蛋白相互作用来影响β-catenin功能的。
2018年,有研究者对欧洲IPH、HPH和厌食药物相关PH患者进行基因测序发现,除了BMPR2(15.3%)、TBX4(1.3%)、ACVRL1(0.9%)、SOX17(0.9%)、ENG(0.6%)、SMAD9(0.4%)和KCNK3(0.4%)外,还发现了新的突变基因——AQP1(0.9%)和ATP13A3(1.1%)。其中AQP1可编码质膜水通道蛋白1,维持血管张力。家系研究发现,在5例HPH患者中发现了AQP1错义突变,即p.R195W和p.V176E[11]。动物实验发现,AQP1基因缺失小鼠内皮细胞迁移和血管生成受损,而应用AQP1抑制剂可减轻缺氧诱导的AQP1表达上调,并通过降低右心室压力、减缓右心室肥厚而逆转肺血管压力升高[45]。ATP13A3是ATP酶家族中的一员,能跨膜转运多种阳离子,在大血管细胞类型中呈高表达[11]。ATP13A3的3个杂合子移码突变会导致ATP酶催化活性丧失,其他杂合子错义突变则会破坏催化结构域的构象,对蛋白功能产生严重影响。研究表明,敲除ATP13A3基因后,内皮细胞增殖减少、凋亡增加[11]。内皮功能障碍介导的抗凋亡细胞不断增殖及其导致的疾病关键部位增殖细胞明显增多,是PH的始动因素[46]。因此,ATP13A3的促细胞增殖作用可能是引起PH的内在关联因素。
6 基于致病基因的靶向治疗
PH患者的异常BMP信号和表观遗传可部分通过诱导Warburg线粒体的非耦合糖酵解代谢,进而为异常细胞提供能量、促进细胞增殖[47]。小分子PDK抑制剂二氯乙酸是针对上述代谢途径的新疗法,其能逆转PASMCs和右心室心肌细胞的Warburg表型。一项Ⅳ期IPH临床研究显示,二氯乙酸能降低IPH患者mPAP和右心室收缩压,延长其6 min步行距离;但携带SIRT3和UCP2基因功能变异的IPH患者对二氯乙酸治疗无应答[48]。
与BMPR2相关的靶向治疗包括补救突变基因功能或通过非突变等位基因产生功能性受体,以增强BMPR2信号传导。补救策略包括使用通读化合物,如Ataluren(PTC-124),其可促进终止密码子通读提前,并从突变等位基因中产生功能性全长BMPR2蛋白[49]。使用化学伴侣也可以补救错义突变,如4-苯基丁酸盐可同时增加错误折叠和促进功能性BMPR2蛋白从细胞内质网转运到细胞表面。其他靶向治疗方法正在开展临床前试验,其一是增强非突变BMPR2蛋白信号:传递重组配体BMP9或使用BMP小分子激动剂FK506,增强BMPR2介导的内皮细胞信号传导,进而逆转PH模型;其二是通过溶酶体抑制剂(如羟氯喹)抑制BMPR2降解,进而预防PH进展。研究表明,靶向敲除小鼠PASMCs上的LRP1基因可使TGF-β信号增强(去抑制),进而诱导PH发生和肺动脉远端肌化;过氧化物酶体增殖物激活受体(peroxisome proliferators-activated receptors,PPAR)γ激动剂吡格列酮能抑制人PASMCs的TGF-β1-CTGF通路激活,抑制PASMCs增殖,进而改善LRP1缺失引起的PH[50]。
7 PH的遗传检测和全球策略
目前,美国和欧洲均在进行大规模的基因学研究。美国国家生物样本数据库(https://www.pahbiobank.org)建立了3 000多例PH患者的生物样本遗传数据,包括靶向DNA测序、全外显子组和全基因组测序等。美国肺血管疾病表型组学计划旨在将PH临床表型与多个“omics”分析结合起来,并在传统PH分类基础上定义新的分子分类。英国BRIDGE项目(https://bridgestudy.medschl.cam.ac.UK/PAH.shtml)已经将PH列为罕见病,目前已对1 250多例欧洲IPH/HPH患者进行潜在遗传突变分析。目前,临床上遗传检测发现的PH相关基因有BMPR2、ACVRL1、ENG、SMAD4、SMAD9、CAV1、KCNK5、KCNK3、BMP9和NOTCH3。遗传检测适用人群包括HPH、IPH、厌食药相关性PH、PVOD/PCH和儿童PH患者[51];需同时对PH患者的亲属进行遗传咨询和遗传检测,对有风险的家庭成员应每3年进行1次教育和随访,以便早期诊断和治疗PH。
8 小结
PH患者存在多基因突变、遗传异质性和不完全外显性等特点,导致其基因学表现复杂,除了目前已知的致病基因突变外,可能还有其他罕见基因突变,同时PH的遗传和环境修饰也有待鉴定。此外,儿童和成人PH相关致病基因可能有所不同。未来,随着基因学及基因组学研究不断深入,PH的表型和靶向治疗研究将迎来新的突破。
作者贡献:王蔚进行文章的构思与设计,文章的可行性分析,撰写、修订论文;钟子晴、荀秋芬、祝国风进行文献/资料收集、整理;杨青负责文章的质量控制及审校,并对文章整体负责、监督管理。
本文无利益冲突。
