耐热型光子晶体结构生色织物的制备及其光学性质研究
2022-11-05王少杰王晓辉李义臣胡敏干邵建中
王少杰,王晓辉,赵 倩,李义臣,胡敏干,邵建中
(1.浙江理工大学生态染整技术教育部工程研究中心,浙江杭州 310018;2.海宁绿盾纺织科技有限公司,浙江嘉兴 314408)
光子晶体(PCs)是一种具有周期性介电结构的材料,最基本的性质是具有光子禁带[1],波长在禁带范围内的电磁波都被禁止传播。当光子禁带落在可见光范围(380~780 nm)内时,与之相对的可见光不能够通过该光子晶体结构而被选择性地反射,进而在周期性排列的光子晶体表面形成相干衍射。相长干涉的反射光刺激人眼,进而产生特定的结构色视觉效果。光子晶体结构生色材料在智能响应、智能显示、防伪、传感、印刷以及纺织品着色[2-7]等众多领域受到广泛关注。
目前常用的光子晶体构筑基元可以分为有机纳米微球和无机纳米微球。有机纳米微球通常有聚苯乙烯(PS)、聚甲基丙烯酸甲酯(PMMA)、聚甲基丙烯酸(PMAA)等纳米微球。无机纳米微球通常有二氧化硅(SiO2)、二氧化钛(TiO2)、氧化亚铜(Cu2O)等纳米微球[8-13]。有机纳米微球具有原材料价格低、制备简便、易于自组装、适合大批量合成和大面积光子晶体结构生色材料制备等诸多优点。然而,有机纳米微球的熔融温度较低,耐热性较差[14],随着温度的升高,大分子链的运动不断加剧,纳米微球的球形度逐渐变差,直至完全熔融而失去固定形状。以纳米微球为结构基元的光子晶体的结构规整性和有序度也随之削弱,直至完全失去规整性。因而,常见的有机纳米微球及其自组装而成的光子晶体的耐热性较差,例如以PS 微球为结构基元的光子晶体在100 ℃下放置1~2 min 就会完全失去结构色,极大地限制了有机纳米微球及其光子晶体的应用。
为了提高光子晶体结构的耐热稳定性,有研究通过合成SiO2、TiO2等无机纳米微球作为光子晶体结构基元,光子晶体的耐热性显著提升,但合成步骤繁琐,合成量和固含量难以提升,也难以实现大面积耐热型光子晶体结构生色材料的制备。因此,发展一种方法简便且适合大量生产耐热型有机纳米微球的制备方法具有重要意义。本实验采用乳液聚合法制备了不同耐热温度的单分散聚苯乙烯纳米微球,并以其作为结构基元,以黑色涤纶织物[15]为基材,采用自组装[16]法制备具有光子晶体结构生色特性的纺织材料。通过改变合成过程中单体与交联剂的比例,实现对光子晶体耐热稳定性及其光学特性的有效调控。
1 实验
1.1 材料与仪器
材料:黑色涤纶织物(210D,市售),苯乙烯(St,分析纯,国药集团化学试剂有限公司),十二烷基硫酸钠(SDS,分析纯,天津市科密欧化学试剂有限公司),过硫酸钾(KPS,分析纯,天津市永大化学试剂有限公司),二乙烯苯(DVB)、二甲苯[分析纯,阿拉丁试剂(上海)有限公司],去离子水(电导率18 MΩ/cm,实验室Milli-Q 纯化)。
仪器:KH-7700 型光学显微镜(美国科视达公司),Nano-S 动态光散射激光粒度仪(英国Malvern 公司),ALTRA55 场发射扫描电镜(德国ZEISS 公司),Maya2000PRO 深紫外光谱仪(美国Ocean Optics 公司),EOS600D 型数码相机(日本佳能公司),Q2000 差示扫描量热仪(美国TA 公司)。
1.2 单分散PS纳米微球的制备
采用乳液聚合法制备聚苯乙烯纳米微球。称量1 050 mL 去离子水、0.75 g SDS 加入装有回流冷凝管、电动搅拌器和氮气入口管的3 000 mL 四口圆底烧瓶中,以360 r/min 搅拌5 min,使乳化剂充分溶解并均匀分布,然后加入总质量为450 g 的St 和DVB,以360 r/min 搅拌均匀,通入氮气驱氧15 min,对体系升温,待温度达到85 ℃后,加入一定量KPS 引发反应,并开始计时。聚合反应全程必须在氮气保护下进行,冷却至室温后出料。
1.3 织物前处理
将黑色涤纶织物超声清洗10 min,采用去离子水冲洗2 次后置于干燥箱内烘干,完全干燥后裁剪为直径5 cm 的圆片状待后续使用。
1.4 PS光子晶体结构生色织物的制备
将圆片状涤纶织物置于直径5 cm 的容器底部,随之将超声分散后具有一定浓度的PS 纳米微球乳液加入织物表面,使织物完全浸没,然后将载有自组装液和涤纶基材的容器置于恒温恒湿箱(温度60 ℃,相对湿度60%)中12 h,待水分完全蒸发后,得到光子晶体结构生色涤纶织物样品。
1.5 测试与表征
1.5.1 粒径及单分散性
将制备所得的纳米微球乳液用去离子水稀释至0.01%,滴入石英比色皿中,采用动态光散射激光粒度仪进行测试。
1.5.2 表面形貌
采用场发射扫描电镜进行观察。
1.5.3 光学性能
采用光学显微镜及数码相机拍摄光子晶体及其制备过程中样品的形貌,采用深紫外光谱仪测试结构生色光子晶体的反射率。
1.5.4 交联度
采用凝胶萃取法测定质量,按照下列公式计算交联度:
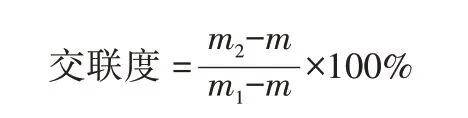
式中:m为装样品前筛网质量;m1为装样品后筛网加样品质量;m2为二甲苯140 ℃萃取5 h 干燥后筛网加样品质量。
1.5.5 玻璃化转变温度
将制备所得纳米微球乳液完全烘干,取2~8 mg,采用差示扫描量热仪进行测试,升温速率10 ℃/min。
2 结果与讨论
2.1 交联型纳米微球的表征
2.1.1 粒径及单分散性
DVB 作为一种带有双官能团的功能性单体,在参与St 聚合反应的过程中既可以起到交联剂的作用,使PS 纳米微球内部形成三维网状的交联结构,也可以作为反应体系中的一种成分,与St 进行共聚。由图1 可看出,随着DVB 用量的增加,所制备的纳米微球粒径呈类线性减小趋势,这是由于随着DVB 用量增加,交联程度提高,纳米微球的结构更为致密。此外,所制备的纳米微球单分散性均小于0.08,表明微球尺寸均一,有望作为光子晶体的优良构筑基元[17]。

图1 DVB 用量对纳米微球粒径及单分散性的影响
2.1.2 SEM
由图2 可以看出,所制备的纳米微球球形度良好,而且大小均一,可以作为结构生色光子晶体的优良构筑基元。

图2 不同DVB 用量所得纳米微球的SEM 照片
2.1.3 交联度
微球交联度随DVB 用量变化见表1。

表1 微球交联度随DVB 用量变化
由表1 可以看出,DVB 用量为4%时,微球的交联度为28.83%,交联程度比较低;DVB 用量为8%时,微球的交联度显著增加到53.87%;DVB 用量继续增加至12%时,微球交联度的提高幅度减小,交联度为65.62%;当DVB 用量进一步增加至16%时,微球的交联度虽有所提高,但是提高幅度进一步减小,交联度为72.39%。可见交联度并非随着交联剂用量的增加呈线性增加。一方面是由于该合成反应控制DVB 和St 总质量不变,DVB 用量是相对St 而言的,因而虽然DVB 用量按4%递增,但是DVB 用量绝对值的增长幅度却在不断减小;另一方面,随着DVB 用量的不断增加,St 的绝对量不断减少,单位量DVB 与St 发生碰撞和反应的概率下降。
2.2 耐热型光子晶体结构生色膜的表征
2.2.1 耐热稳定性
为分析和评价交联型纳米微球的耐热性能,将不同交联程度的纳米微球作为自组装基元,在涤纶织物表面通过重力自组装构建光子晶体结构,然后将光子晶体结构生色织物样品置于不同温度下观察其结构色的变化。
由图3 可以看出,使用未交联PS 纳米微球作为自组装基元构筑的光子晶体结构生色织物,80~100 ℃处理1 min 时,结构色出现明显变化,100 ℃处理1 min 时,结构色完全消失;当DVB 用量为4%时,80~100 ℃处理1 min 后结构色基本无变化,110 ℃处理1 min 后,结构色发生一些红移,120 ℃处理后,结构色则完全消失,可见耐热性有所提高;DVB 用量继续升高至8%~16%时,80~130 ℃处理1 min后,结构色基本无变化,140 ℃处理1 min 后,结构色出现轻微变化,150 ℃处理后结构色效果变化明显,但未完全消失,说明光子晶体结构的耐热稳定性明显提升。这是由于随着交联程度的提高,PS 纳米微球内部逐步形成致密的三维网状交联结构,即使温度升高,大分子链也难以大幅度自由运动,因而微球的形状得以保持,以该类交联型微球作为结构基元所构建的光子晶体结构也得以稳定。
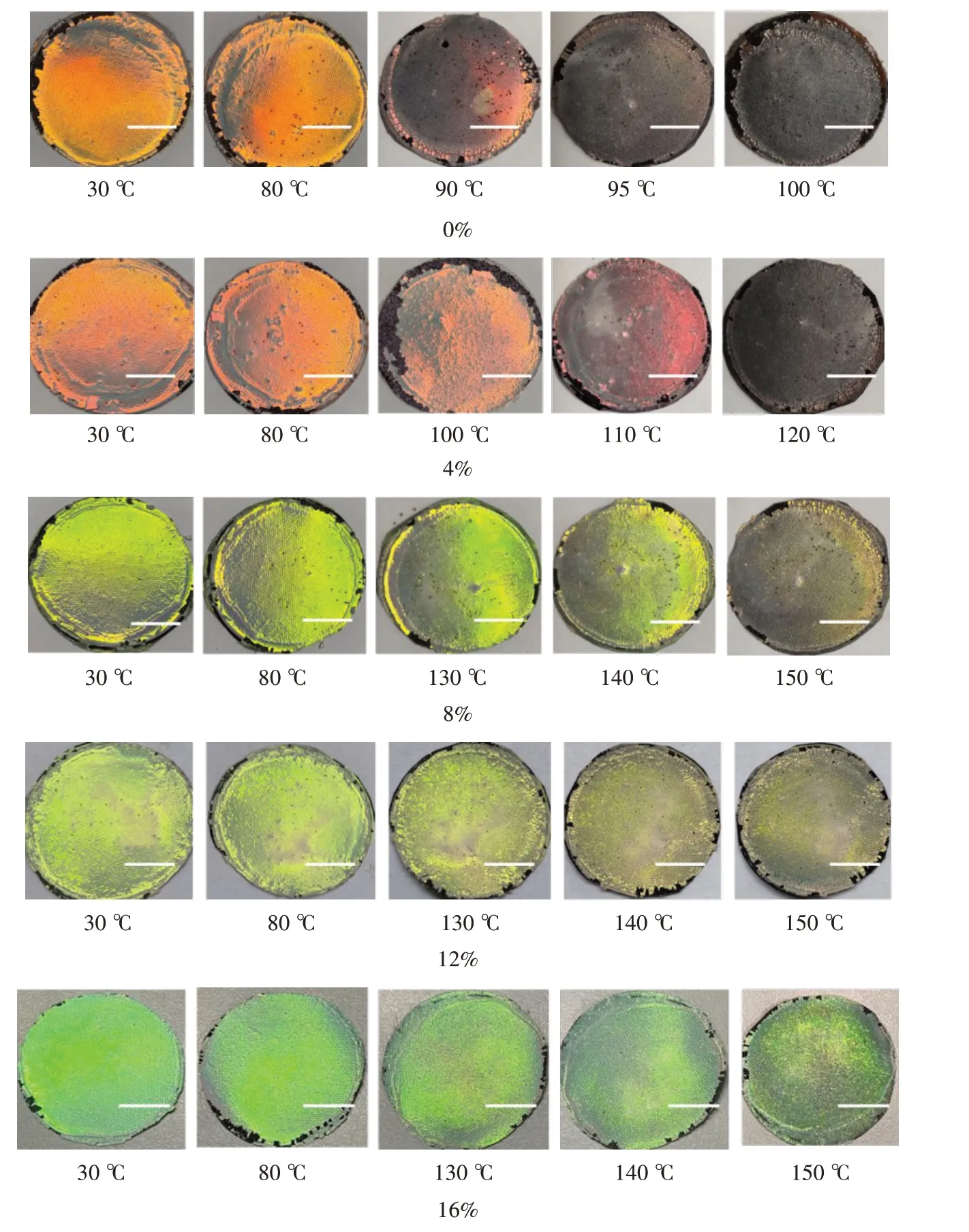
图3 不同DVB 用量微球自组装光子晶体在不同温度下的结构色变化
为了进一步探究结构色在高温条件下变化的原因,对光子晶体的微观结构进行电子显微镜观察分析。由图4 可以看出,以未添加交联剂的PS 纳米微球作为自组装基元所制备的光子晶体,80 ℃处理1 min时,纳米微球结构排列规整,微球间存在明显空隙;90 ℃处理时,纳米微球熔融变形并且产生粘连现象,导致微球空隙间的部分空气被熔融物所取代,进而使其有效折光指数升高,结构色红移(可以由下面公式推导);当温度升至100 ℃时,纳米微球完全粘连在一起,以致光子晶体内部的空气被完全熔融的PS 所取代,光子晶体结构的折光指数差为0,光子禁带消失,结构色也消失,这也与图3 中结构色消失的现象相吻合。

图4 未交联微球自组装光子晶体在不同温度下处理后的SEM 图像
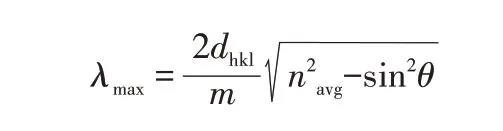
式中:λmax为光子晶体结构色反射率曲线峰值所对应的波长(光子禁带位置);m为衍射能级;dhkl为光子晶体的晶面间距(与微球粒径相关);navg为光子晶体的平均折光指数;θ为入射光的入射角。由公式可知,随着navg增加,λmax相应变大,结构色出现红移。
与图4 形成鲜明对比的是,以交联剂用量16%的PS 纳米微球作为基元自组装所得的光子晶体结构,在80、130 ℃下处理时并未出现明显的熔融粘连现象,在150 ℃下处理时也只发生了轻微的熔融变形(如图5 所示),这也是其在高温条件下仍然可以保持鲜艳亮丽结构色彩的原因。

图5 16%DVB 微球自组装光子晶体在不同温度下处理后的SEM 图像
为探明交联后纳米微球热稳定性提升的根本原因,对其玻璃化转变温度(Tg)进行研究。由图6 可知,随着DVB 用量的不断提高,制备的PS 纳米微球的玻璃化转变温度(Tg)由初始的96.8 ℃升高至116.9 ℃。这是由于纯St 单体聚合而成的纳米微球是由较长的聚苯乙烯高分子链卷曲缠绕而成,高分子链上无极性基团,分子链间除范德华力外不存在其他分子间作用力,虽然主链存在大量刚性的苯环,但是当温度升高后分子链仍容易发生运动;引入DVB 参与反应后,高分子长链间会产生共价键合,极大地限制了高分子链的自由运动,因此Tg升高。

图6 不同DVB 用量微球的玻璃化转变温度
2.2.2 光学性能
由图7 可以看出,以未交联PS 纳米微球自组装形成的光子晶体结构,80 ℃处理2 min 内结构色无变化,而将时间延长至4 min 或者更长时结构色明显变暗;90 ℃处理1 min 时结构色就明显变暗,延长至6 min 时结构色完全消失;而当温度升高至100 ℃时,处理1 min就足以使结构色完全消失。

图7 未交联微球自组装的光子晶体在不同温度下处理不同时间的结构色变化
图8a~图8c 展示的反射率曲线与图7 光子晶体结构色变化趋势一致。由图8a、图8d 可以看出,在80 ℃条件下,随着处理时间逐渐延长,反射强度逐渐降低;90 ℃处理1 min 后,反射强度大幅度下降,处理6 min 后反射强度基本为0,表明结构色完全消失(如图8b、图8d 所示);而100 ℃处理1 min 后,反射强度降为0(如图8c、图8d所示)。
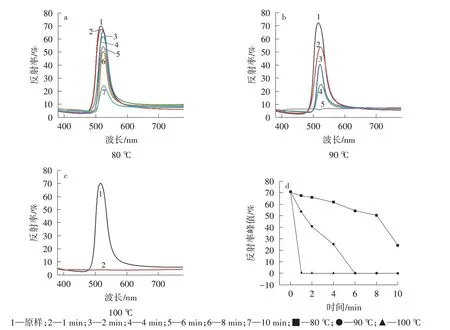
图8 未交联微球自组装光子晶体在不同温度下处理不同时间的反射率曲线以及反射率峰值随处理时间的变化趋势
图9 为PS 纳米微球自组装所得光子晶体在不同温度下处理不同时间的结构色变化(因为在80~100 ℃下纳米微球自组装形成的光子晶体颜色随着时间延长基本无变化,所以选择110 ℃开始讨论)。

图9 72%交联度微球自组装的光子晶体在不同温度下处理不同时间的结构色变化
由图9 可知,纳米微球构建的光子晶体110 ℃处理1~10 min 时,结构色均未发生明显变化;120 ℃处理时,结构色随着处理时间的延长发生些许变化,处理8 min 时结构色开始发生较明显的变化;130 ℃处理1 min 时结构色明显变暗,当时间延长至10 min时,与原样相比结构色亮度骤减;140 ℃处理1 min时,结构色发生较大变化,亮度明显降低,但当处理时间延长至10 min时,仍具有颜色。
图10a~图10d 展示的反射率曲线与图9 光子晶体结构色变化趋势一致。由图10a、图10e 可知,在110 ℃条件下处理反射率峰值基本无变化;120 ℃处理6 min 时,反射率峰值降低,时间延长至8~10 min,反射率峰值较大幅度下降(见图10b、图10e);130、140 ℃处理1 min 时,反射率峰值大幅度下降,时间延长至10 min 时,反射率峰值未完全消失,表明结构色未完全消失(见图10c、图10d、图10e)。

图10 72%交联度微球自组装的光子晶体在不同温度下处理不同时间的反射率曲线及反射率峰值随处理时间的变化趋势
3 结论
(1)DVB 在反应过程中主要起交联作用,而非共聚作用。随着DVB 用量的增加,PS 纳米微球的交联度提高,玻璃化转变温度也升高(由初始的96.8 ℃升高至116.9 ℃)。
(2)未交联纳米微球自组装的光子晶体100 ℃处理1 min 时,结构色完全消失,光子晶体结构基元熔融粘连,微球间空隙被完全填充。DVB 用量为8%~16%时,微球交联度均超过50%,自组装形成的光子晶体150 ℃处理1 min 时,微球间的空隙仍基本保留,结构色仍然明显可见。
(3)未交联纳米微球自组装的光子晶体90 ℃处理6 min、100 ℃处理1 min 时,结构色完全消失,反射峰强度几乎为0;交联度为72%的纳米微球自组装的光子晶体120 ℃处理6 min 时,结构色仍未发生明显变化,140 ℃处理10 min 时,光子晶体结构生色织物仍具有颜色,反射光谱变化与之吻合。
