小麦酵母双杂交文库构建及文库质量的鉴定
2022-11-02李立群王冰心杨智全
李立群,王冰心,吕 千,杨智全
(西北农林科技大学 农学院,陕西杨凌 712100)
构建高质量的酵母双杂交文库是研究蛋白互作关系,筛选未知基因的基础[1-2],是揭示基因蛋白功能及解析其作用机理的前提。近年来,酵母双杂文库被广泛应用,在挖掘新基因及基因功能研究方面发挥了重要作用。农作物如水稻、棉花、青稞[3-5]及麦类作物[6-8]中普遍采用,水果[9-10]蔬菜[11-13]也屡见报导。杨少丽等[14]利用Gateway技术构建菊芋块茎全长cDNA文库,用于菊芋功能基因组研究、新基因筛选、高通量EST测序以及菊芋cDNA芯片制备。叶夏林等[15]构建南通小方柿cDNA文库,筛选Dk GA2ox2基因上游转录因子,研究矮化基因调控机制。陈玉婷等[16]构建花生受青枯菌诱导的根部组织文库,筛选到抗青枯病Ah RRS5基因的互作蛋白AhSBT1.6,并证明其参与调控花生青枯病抗性。可见,酵母双杂文库在蛋白互作及功能研究中发挥了不可替代的作用。
小麦是中国主要的粮食作物,粒质量是重要的产量性状,粒质量基因研究国内外报道较多[17-19],小麦育种技术研究与新品种培育创新团队已经取得了一定成果[20-22]。然而,为了深入开展基因功能研究,揭示调控籽粒大小的分子机制,筛选粒质量基因作用的关键靶标蛋白,必须首先构建基因文库。目前在小麦中建立的cDNA文库多基于SMART技术,以小麦叶片为主要材料,集中在抗病基因的挖掘和研究[23-25]。近期王巧慧等[26]以小麦抗病材料叶片构建酵母双杂cDNA文库,筛选获得2个与热休克蛋白HSP40-70互作的蛋白质,为抗逆研究奠定基础。但基于gateway技术以中国春小麦各种组织为材料构建酵母双杂交文库尚未见报道。
本研究中,以中国春小麦不同组织及不同花后时间的籽粒为材料,基于Invitrogen的gateway技术,利用Clone Miner II试剂盒构建中国春小麦cDNA初级文库,然后LR重组构建出次级文库,再依据次级文库质粒构建出酵母双杂交文库,并以小麦粒质量基因TaDAR1(GeneID:AK457429.1)为例,构建诱饵表达载体PGBKT7-TaDAR1,然后用mating法进行文库筛选,并对文库质量及有效性进行鉴定。研究为初学者了解酵母双杂文库构建、酵母感受态细胞制备以及从文库中筛选基因的方法提供参考。
1 材料与方法
1.1 材料
1.1.1 试验材料 中国春小麦品种,生物技术育种研究实验室繁殖保存。2019播种于西北农林科技大学北校区小麦育种试验田,在小麦孕穗期,分别取叶片、茎杆组织及幼穗,-80℃保存,待小麦开花后再分别取3、6、9、12、15、20、25 d的小麦籽粒,液氮速冻,-80℃保存,备用。
1.1.2 主要试剂 载体:p DONR222、p GADT7、p GBKT7;菌株:酵母Y2H、Y187、大肠杆菌等。Invitrogen公司试剂盒:Clone Miner II cDNA Library Construction Kit、TRIzol Reagent、Fast-Track MAG m RNA isolation Kit、SuperScript III First-Strand Synthesis System for RT-PCR、BP ClonaseII enzyme mix及LR Clonase II Mix等;酵母质粒提取试剂盒、OXOID公司SOC YNB、Yeast extract、北京天根公司胶回收试剂盒、质粒小提试剂盒、诺唯赞生物科技公司各种限制性内切酶、2XTaqMaster Mix、Clon Express Entry One Step Cloning Kit。本试验所用的引物由北京擎科生物科技公司西安分公司和生工生物(上海)技术有限公司合成,所有的测序工作由杨凌奥科生物科技有限公司承担。
1.2 方法
1.2.1 中国春小麦总RNA提取及m RNA分离取前期冻存的中国春小麦叶片、幼茎、幼穗,以及花后不同发育时期的籽粒,采取Trizol法提取各组织总RNA,用微量核酸分析仪nanodrop2000检测总RNA的质量和浓度,配制10 g/L的琼脂糖凝胶电泳检测RNA的完整性,然后根据RNA的量进行等比例混合建库。
m RNA的分离纯化,具体参照Oligotex m RNA Midi Kit说明书进行。取10μL进行电泳质检,观察电泳条带是否呈弥散状,以及高亮度所在区域。
1.2.2 中国春小麦cDNA文库的构建 (1)中国春小麦初级文库的构建:将分离纯化好的m RNA反转录合成双链cDNA,将cDNA分级分离和收集后用于BP重组反应。反应体系为20μL,包 括13μL cDNA,2μL p DONR222(200 ng/μL),5μL BP ClonaseII enzyme mix。反应产物经电转化大肠杆菌DH10B感受态细胞后,加入SOC培养基,置于37℃、220 r/min摇床培养1 h。培养结束后,涂布平板用于库容量鉴定,剩余培养物加入甘油至终浓度20%,于-80℃保存,即为中国春小麦cDNA初级文库菌液。
(2)中国春小麦cDNA次级文库的构建:抽提中国春小麦cDNA初级文库质粒,稀释到300 ng/μL,利用LR重组反应构建中国春小麦cDNA次级文库。反应体系为20μL,其中初级文库质粒(300 ng/μL)1μL,pGADT7-DEST(300 ng/μL)1μL,LR Clonase II Mix 4μL,dd H2O 14μL补足体积,将体系混匀后25℃反应18~22 h。反应结束后将10μL重组产物和100μL电转感受态细胞冰上混匀,在1 500 V、200Ω、25μF电击条件下,转化DH10B感受态细胞。将电转化产物移入新的离心管,培养后涂布平板用于库容量鉴定,剩余培养物加入甘油至终浓度20%,存于-80℃,即为中国春小麦cDNA次级文库菌液。
(3)中国春小麦cDNA文库质量鉴定:取转化后细菌原液10μL稀释1 000倍后,从中取出50μL涂布LB平板(初级文库抗性为卡那霉素,次级文库抗性为氨苄青霉素),37℃,过夜培养,第2天计数。每毫升大肠杆菌文库菌液库容量(CFU/m L)=平板上的克隆数/平板上涂布菌液的体积(μL)×n×1 000(μL),n为稀释倍数。
(4)重组率和插入片段长度的分析鉴定:重组率是菌落PCR中所有阳性插入片段的克隆在总检测克隆数中所占的比例;插入片段平均长度则是随机菌落PCR所鉴定得到的插入片段长度的总和的平均值,本试验需要通过文库质粒转化和克隆鉴定来获得。操作步骤如下:取200μL感受态宿主菌(DH5α),放置冰上融化,加入1μL文库质粒,轻轻混匀后置于冰上30 min,42℃水浴热休克90 s,迅速放入冰上2 min,加入300μL SOC培养基(不含抗生素),置于37℃摇床,转速200 r/min,复苏45 min,菌液分别稀释1 000倍、10 000倍涂布于15 cm培养皿上,37℃过夜。然后,挑单克隆做菌落PCR,扩增反应所需引物见表1,扩增程序见表2。PCR产物用10 g/L琼脂糖凝胶电泳检测,以此来计算初级和次级文库重组率及插入片段长度。

表1 所用引物Table 1 Primers used in this study
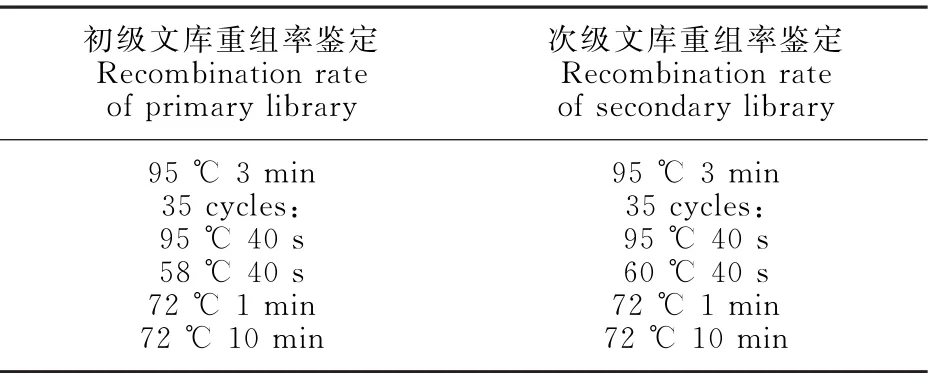
表2 文库鉴定PCR程序Table 2 PCR reaction conditions for identification of library
1.2.3 酵母双杂交文库的构建 (1)酵母感受态细胞的制备及转化。酵母感受态细胞制备:取-80℃保存的酵母(Y187或Y2HGold)菌株,在超净台划线于含卡那霉素抗性的YPDA培养基上进行活化培养,30℃培养4~5 d。当有肉眼可见的菌落时,挑取单菌落(直径2~3 mm)于15 m L离心管中,30℃,160~180 r/min振荡培养16~20 h。取5μL培养物到250 m L三角瓶中,加入50 m L YPDA培养基中,30℃下170 r/min培养18~22 h,至OD600值达到0.3左右。6 000 r/min离心6 min,弃上清,用100 m L YPDA重悬浮菌体,30℃振荡培养5 h至OD600达到0.5左右。然后将100 m L酵母菌液分装为2管,6 000 r/min离心6 min,弃上清,每管均加入40 m L无菌的dd H2O,悬浮沉淀,6 000 r/min离心5 min,再重复一次,去尽上清。每管用1.5 m L现配的1.1×TE/LiAc溶液悬浮菌体,转移至2 m L离心管,13 000 r/min离心25 s弃上清,每管加入1.1×TE/LiAc 600μL重悬菌体,即完成感受态细胞的制作。
中国春小麦cDNA次级文库质粒转化酵母感受态Y187:在15 m L无菌的离心管中依次加入次级文库质粒5μg、已变性鲑鱼精20μL、酵母感受态细胞600~1 200μL、1×PEG/LiAC 2 500 μL,轻轻混合。然后金属浴中30℃孵育45 min,其间颠倒混匀3次。在超净台中加入160μL的DMSO之后轻轻混匀,在42℃金属浴中孵育20 min,其间轻轻上下颠倒混匀一次。酵母转化完成,将离心管室温下6 000 r/min、离心6 min、弃上清,用1 m L YPDplus重悬细胞,5 000 r/min离心5 min,弃尽上清,再用15 m L质量分数为0.9% NaCl悬浮细胞以保持细胞正常渗透压,将菌液均匀涂布于70~100个15 cm含有SD/-Leu营养缺陷培养基上,30℃,倒置培养4~5 d至菌落长出。
(2)酵母双杂文库的鉴定:从Y187菌液中吸取150μL酵母菌液,分别稀释10、100、1 000倍,吸取100μL将不同浓度的菌液均匀涂布于15 cm,SD/-Leu营养缺陷培养基平板上,30℃,倒置培养4~5 d,待白色菌斑长出,计数并计算文库转化效率。酵母文库转化效率(CFU/μg)=(CFU×悬浮液体积×稀释倍数)/[初始涂布体积×转化DNA数(μg)]。
待菌落长出后,将涂有Y187菌株的SD/-Leu平板放置在4℃冰箱3~4 h,每平板加5 m L含30%甘油的YPDA溶液,将菌落全部刮下,收集到500 m L三角瓶中,估计酵母细胞数目。如果细胞数目低于2×107CFU/m L,浓缩后分装1 m L/每管,-80℃分装保存,即为“mating”文库菌。
1.2.4 文库的筛选及回转验证 (1)诱饵菌株构建:本试验使用同源重组的方法构建BD诱饵表达载体。其中,插入BD载体的酶切位点为BamHⅠ和EcoRⅠ,构建诱饵载体所需引物见表1。使用菌落PCR鉴定阳性单菌落,并提取质粒进行酶切,以检测诱饵载体的正确性,然后将BD诱饵表达载体转化Y2H酵母菌株,并涂布于SD/-Trp营养缺陷平板上。30℃倒置培养4~5 d,即为含有BD诱饵表达载体的菌株。
(2)诱饵菌株的自激活和毒性验证:分别挑取含有BD诱饵表达载体及含有BD空载体的Y2 H单菌落,溶于10μL质量分数为0.9% NaCl溶液中,各吸取1μL菌液,点样在缺体培养基SD/-Trp(一缺)及SD/-Trp-His-Ade(三缺)上,30℃培养4~5 d,观察酵母菌的变化。如果酵母菌斑能够在2种缺体培养基平板上生长,说明诱饵蛋白具有自激活效应,如果只能在一缺培养基上生长,不能在三缺培养基上生长,则说明无自激活活性。
分别挑取含有BD诱饵表达载体及含有BD空载体的Y2 H单菌落,将其接种于50 m L含有50μg/m L卡那霉素的SD/-Trp液体培养基中,在30℃、250 r/min的条件下震荡培养18~20 h,并且比较两者的OD600值,从而确定诱饵蛋白的表达是否对酵母细胞有毒性。如果无毒性,含有BD诱饵表达载体Y2H菌株的生长速度与含有BD空载体Y2 H菌株的生长速度相近。如果有毒性,含有BD诱饵表达载体Y2H菌株的生长会受到抑制,其OD600值小于含有BD空载体Y2H菌 株OD值。
(3)文库的筛选:参考Clontech酵母双杂文库说明书,采用“mating”的方法进行文库筛选。当文库菌与诱饵菌杂交后,显微镜下可观察到酵母合子细胞,即视野中出现米老鼠头像或者三叶草图像,说明酵母杂交成功。常温下5 000 r/min离心8 min收菌,用质量分数为0.9% NaCl重悬菌体,涂四缺平板培养基SD/-Trp/-Leu/-Ade/-His,30℃倒置培养5 d,观察酵母生长情况。挑取四缺平板上的酵母菌斑,点在含有40μg/m L X-α-gal和200 ng/m L Ab A的四缺平板上,若酵母能正常生长且变蓝,即为酵母双杂交阳性菌。
(4)回转验证:使用酵母质粒提取试剂盒提取酵母双杂交阳性菌的质粒,转化大肠杆菌(DH5α感受态),根据Amp的抗性分离质粒得到只转入AD-pery载体的菌斑,抽提质粒并测序,通过比对NCBI数据库初步确定可能的互作基因。从中国春小麦c DNA中获取该基因的全长序列,并将其接入AD载体。
再将连接互作基因全长的AD质粒和诱饵质粒一起回转到Y2H酵母菌中,将转化的菌株涂在DDO二缺平板及QDO四缺平板,观察它在30℃、3~5 d内是否生长。假如在2种营养缺陷型平板均有生长,说明下游报告基因被激活,也就证明转入的2种片段之间存在互作关系;假如只在二缺平板上生长,说明报告基因没有被激活,二者没有互作关系。
2 结果与分析
2.1 RNA的提取及mRNA检测
采用Trizol法分别提取中国春小麦叶片、幼茎、幼穗和花后不同时期嫩籽粒的总RNA,电泳检测条带清晰,28S与18S的亮度比接近2∶1,说明总RNA完整性良好(图1-A)。m RNA分离纯化后,电泳质检得到弥散状条带,且弥散状高亮度区集中在大分子量区域1 000 kb以上(图1-B),符合构建文库的要求。
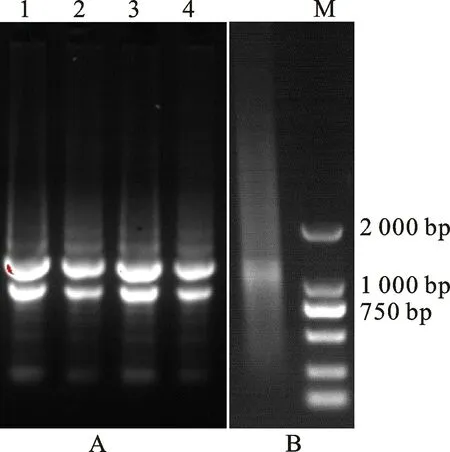
图1 中国春小麦总RNA和mRNA的电泳图Fig.1 Agarose gel electrophoresis of total RNA and isolated RNA of Chinese Spring wheat
2.2 初级文库的构建及鉴定
将纯化后的m RNA反转录成cDNA后,进行BP重组反应。将反应物电转至DH10B获得中国春小麦cDNA初级文库菌液。取10μL初级文库菌液稀释1 000倍,从中取50μL涂布含有卡那霉素的LB平板,37℃过夜培养,平板上的克隆数为912(图2),(平板上的克隆数/50μL)×稀释倍数×1 000μL=912/50μL×1 000×1 000 μL=1.82×107CFU/m L,即大肠杆菌文库菌液库容量为1.82×107CFU/m L。

图2 初级文库平板鉴定库容量图(克隆数912)Fig.2 Identification of primary library total clones(912 clones)
抽提初级文库质粒,并转化DH5α。随即挑选24个单克隆经PCR检测,结果由图3可知,插入片段平均长度在1 kb以上,重组率大于96%,符合初级文库的质量需求。

图3 初级文库插入片段重组率鉴定图Fig.3 PCR identification of primary library
2.3 次级文库的构建及质量鉴定
将抽提的初级文库质粒经LR重组反应后,电转化DH10B获得次级文库菌液。取10μL次级文库菌液稀释1 000倍涂布含有Amp抗性的LB平板。结果如图4所示,平板克隆数为956,经计算,次级文库容量为(平板上的克隆数/50 μL)×稀 释 倍 数×1 000μL=956/50μL×1 000×1 000μL=1.91×107CFU/m L。

图4 次级文库平板鉴定库容量图(克隆数956)Fig.4 Identification of secondary library total clones(956 clones)
抽提次级文库菌液并转化DH5α,由菌落PCR结果(图5),可见插入片段大小都在1 kb以上,重组率>96%,达到次级文库的构建标准。由此可见,经BP重组反应及LR重组反应构建的中国春小麦cDNA文库连接效率高,库容量大。

图5 次级文库插入片段重组率鉴定图Fig.5 PCR identification of secondary library
2.4 酵母双杂文库的鉴定
将中国春小麦c DNA次级文库转化Y187菌株,获得酵母双杂交文库。本试验共转化次级文库质粒6μg,总悬浮体积11.5 m L,稀释100倍后,取100μL涂平板统计长出的平均酵母菌克隆数为103,计算得酵母文库转化效率约为1.97×105CFU/μg,超过“mating”酵母文库转化效率标准(1×105CFU/μg)。
吸取浓缩后的100μL酵母双杂交文库菌液,稀释10 000倍,涂平板培养长出平均为204个克隆,经计算得酵母双杂交文库容量为2.04×107CFU/m L,超过mating酵母文库标准(1×107CFU/m L)。
2.5 文库的筛选及回转验证
2.5.1 TaDAR1诱饵载体的构建 经过对菌液PCR鉴定的阳性克隆进行测序,序列分析结果与粒质量基因TaDAR1的序列完全一致,同时质粒提取酶切验证结果与预期结果一致,酶切后各片段大小与预期相符(图6)。表明诱饵表达载体构建正确,符合酵母双杂交文库筛选的要求,能够进行后续研究。

图6 诱饵载体酶切后电泳图Fig.6 Fragments of vector after enzyme digestion
2.5.2 诱饵菌株自激活与毒性验证 由图7可知,全长的TaDAR1有自激活活性,因此根据TaDAR1蛋白结构域将其截断为N端和C端两部分,并分别构建BD-TaDAR1-N和BD-Ta-DAR1-C载体。转化Y2H菌株后发现,BD-Ta-DAR1-N无自激活活性,表明TaDAR1激活域在蛋白的C端。因此选择BD-TaDAR1-N作为诱饵载体进行后续试验。
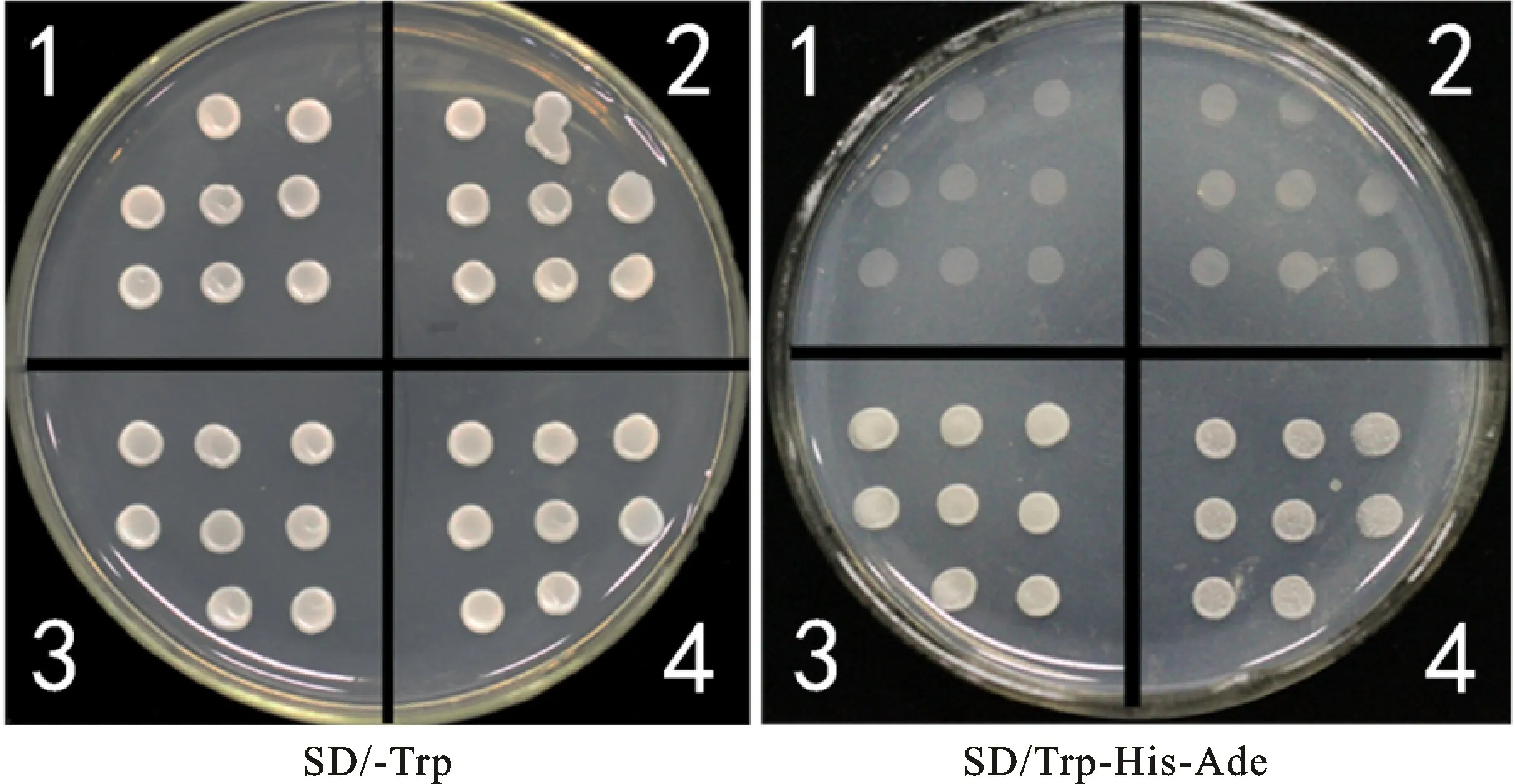
图7 诱饵表达载体酵母Y2H自激活验证图Fig.7 Self-activation detection of bait vector
同时,发现转化诱饵质粒BD-TaDAR1-N的Y2H菌液与转化BD空载菌液的生长速度相近,说明诱饵载体对酵母细胞没有毒性,不会影响文库的筛选。
2.5.3 文库筛选 诱饵菌与文库菌mating后显微镜下观察,有典型的三叶草图形,即酵母合子细胞出现(图8),表明酵母杂交成功,可进行后续试验。
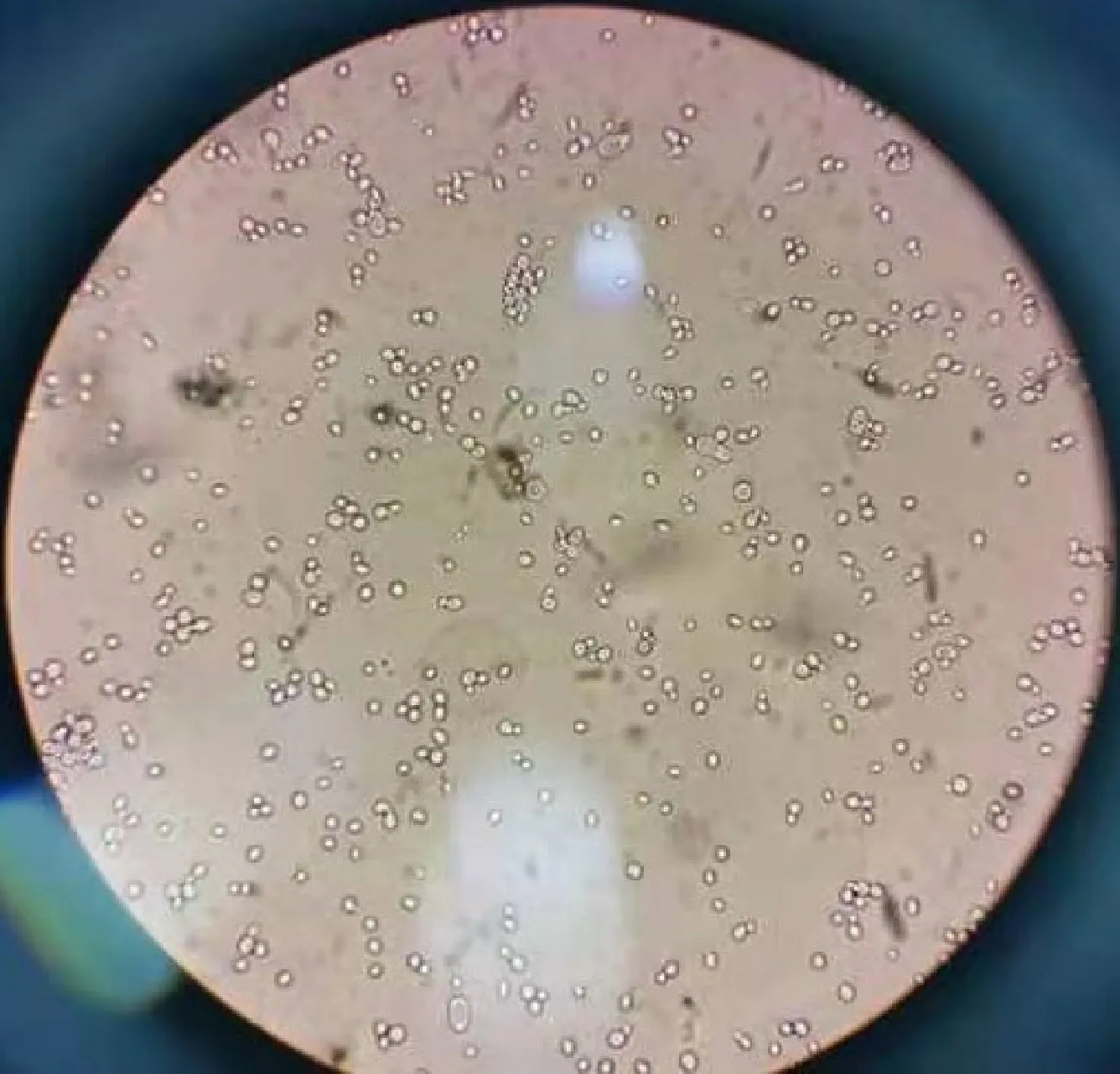
图8 酵母杂交细胞观察Fig.8 Observation of yeast zygote
2.5.4 回转验证 本研究随机挑选3个阳性克隆,抽提质粒后测序,发现其中一个编码蛋白提前终止,为无效片段;另外两个阳性克隆测序序列一致,经比对数据库(https://www.ncbi.nlm.nih.gov/,https://plants.ensembl.org/)后,将其命名为TaDAR1IP(TraesCS3B02G548700.1),该氨基酸全长329 aa,在蛋白N端含有Myb_DNAbind_3结构域(图9)。

图9 TaDAR1IP蛋白序列示意图Fig.9 Sequence of TaDAR1IP protein
构建AD-TaDAR1IP载体同BD-TaDAR1-N回转Y2H菌株,结果含有全长的TaDAR1IP与TaDAR1-N的菌株在DDO和QDO培养基上生长良好,在QDO/X/A长出蓝斑(图10)。结果说明TaDAR1与TaDAR1IP确实存在相互作用,进一步证明利用本方法构建的酵母双杂交文库切实有效。

图10 TaDAR1 IP与TaDAR1互作示意图Fig.10 Interaction validation of TaDAR1 and its candidate protein TaDAR1IP
3 讨论
构建高质量的酵母双杂交文库是大规模筛选候选互作蛋白的基础[27]。本研究提取了中国春小麦叶片,幼茎、幼穗及花后不同发育时期籽粒的总RNA,并将其混合后用于构建酵母双杂交c DNA文库,有效地防止了文库中组织特异性表达基因的丢失[14,28]。
彭晓珏等[29]研究表明利用gateway技术方法构建cDNA文库,不需要传统的酶解和连接过程,能够较大程度地避免低拷贝数基因克隆的丢失,保证了文库的质量和忠实性。王舟等[30]、李晨等[31]研究证明gateway位点特异性重组技术作用显著,通过BP高效重组技术,可以保证材料中m RNA的原始丰度在文库中得到真实的反映,通过LR反应,可以迅速地将目的基因定向重组到各种表达系统如酵母、细菌中,可以高效准确进行基因的表达分析。该方法克服了传统技术中难获得大片段克隆的问题,具有无可比拟的优势。虽然成本稍高,但是更适用于基因组庞大复杂的小麦等作物应用。本研究构建的小麦cDNA文库具有107CFU/m L库容,插入片段平均长度在1 000 bp以上,表明文库覆盖度高且完整性好,文库构建成功。构建的中国春小麦酵母双杂交文库,转化效率为1.97×105CFU/μg,文库容量2.04×107CFU/m L,达到小麦酵母双杂文库的较高要求,可以最大程度地筛选到所有的与靶蛋白具有相互作用的蛋白质分子。
方玉楷等[32]已证明mating法筛选互作蛋白比常用的共转化法效率高。本实验室利用本研究构建的中国春小麦酵母双杂文库已经筛选获得8个抗逆基因和一个粒质量基因的互作蛋白,2个已通过功能验证[33-34],其余基因功能正在进行研究。由此可见本研究构建的中国春小麦cDNA文库质量良好。
使用mating法筛选酵母双杂交文库,可以筛选出与“诱饵蛋白”互作的蛋白[35]。但有时候某些基因诱饵蛋白有自激活作用,会导致假阳性的存在,致使结果不准确[36]。针对此问题,进行文库筛选前,要对重组质粒转入酵母感受态细胞中有无自激活活性进行检测。如果诱饵载体无自激活现象,可直接应用于文库筛选互作蛋白;如果有自激活现象,必须对基因进行截断检测,确定不包含激活的区域,然后才能作为诱饵进行文库筛选,本研究就是一个典型的范例。另一方面,为排除假阳性带来的试验干扰,保证试验结果的真实、严谨,还必须结合BiFC、GST Pull-down、CoIP等试验技术来进一步验证基因的互作关系。
4 结论
本研究以中国春小麦叶、茎、幼穗及花后不同时期的籽粒为材料,利用Gateway技术成功构建了中国春小麦酵母双杂交文库,库容大覆盖面广、质量高。通过mating法筛选互作蛋白,筛选效率高。通过小麦粒质量基因TaDAR1互作蛋白的筛选和验证,表明构建的酵母文库是高质量的、有效的。研究为初学者构建酵母双杂文库及使用“mating”法筛选酵母文库提供借鉴。同时,文库的建立也为后续小麦基因功能的研究奠定了基础。
