马铃薯品种(系)田间晚疫病抗性评价和全基因组遗传多样性分析
2022-10-28李晓川王朝海周平马维吴瑞宋治豪梅艳
李晓川,王朝海,周平,马维,吴瑞,宋治豪,梅艳
毕节市农业科学研究所,贵州毕节 551700
0 引言
【研究意义】晚疫病由致病疫霉菌((Mont.)de Bary)引起,是马铃薯(L.)生产的第一大病害。贵州省所处的乌蒙山区由于气候冷凉潮湿,易大规模爆发晚疫病。致病疫霉菌存在高度的变异性,简单的单个抗病基因导入栽培种所带来的晚疫病抗性,很容易被变异的致病疫霉菌克服。因此,研究马铃薯种质的遗传多样性及抗病种质基因组中可能影响抗性的遗传区段,对马铃薯晚疫病抗性种质创新与利用具有重要意义。【前人研究进展】关于马铃薯晚疫病抗性育种的研究,多集中在克隆具有晚疫病致病疫霉菌小种专化抗性的R(Resistance)基因上,目前,已克隆获得多个R基因,均是NBS-LRR类基因。但无论是具有专一的小种专化抗性的R基因(、、和),还是具有广谱的小种专化抗性的R基因,将单个抗病基因导入栽培种后所具有的晚疫病抗性,在商业化种植后,均易被变异的致病疫霉菌克服。如,将广谱抗性RB基因导入培育的栽培种Biogold,田间种植1年后,便被晚疫病菌克服。马铃薯晚疫病的田间抗性被认为具有相对的持久性,具有非小种专化性抗性,其形成的遗传学基础是由多个基因控制,认为多个R基因的堆叠可以增强马铃薯晚疫病田间抗性表现。【本研究切入点】西南乌蒙山区是中国马铃薯晚疫病发病最为严重的地区,而利用此环境条件,对马铃薯种质资源进行田间晚疫病抗性评价,以及随后进行全基因组遗传多样性分析的研究相对较少。【拟解决的关键问题】本研究通过在晚疫病多发且发病严重地区对马铃薯种质进行田间晚疫病抗性鉴定,获得抗病种质资源,运用现代高通量测序技术分析种质的遗传多样性及抗病种质基因组内可能影响抗性的遗传区段,为马铃薯晚疫病抗性种质创新与利用提供理论依据。
1 材料与方法
1.1 马铃薯田间晚疫病抗性材料筛选及取样
参加马铃薯田间晚疫病抗性筛选的材料,包括自家选育的中高代品系432份以及从各地搜集的马铃薯品种(系)458份(电子附表1)。于2018—2021年分别在贵州省毕节市威宁县草海镇郑家营村、卯关村,威宁县雪山镇团岗村、安家营村、法地村以及七星关区朱昌镇王家冲村内多点种植,每个品种(系)种植50株,种植过程不喷洒晚疫病防治药剂,进行田间晚疫病抗性鉴定。抗性材料筛选:以在晚疫病爆发一个月后,植株依然能够存活的马铃薯品种(系)为抗病材料(贵州毕节一般在3月中下旬种植马铃薯,4月中下旬出苗,6月中下旬晚疫病爆发,7月中下旬可达到当地主栽中晚熟马铃薯品种生长期90—100 d的要求);以在发病10 d左右大部分植株死亡的马铃薯品种为感病材料;其余为半抗病材料。挑选待测序感病材料时,参考欧洲马铃薯栽培种数据库(http://www.europotato.org/quick_search.php)关于晚疫病抗性及其他农艺性状的数据,同时参考马铃薯系谱数据库(http://www.plantbreeding.wur.nl/potatopedigree/index.html)的系谱学数据。取晚疫病抗感材料叶片,分别进行混合,将样品经液氮速冻后保存备用。
1.2 dd-RAD简化基因组测序
将样品送派森诺生物科技公司构建 dd-RAD(double digest restriction site-associated DNA sequencing)简化基因组测序文库。经Illumina NovaSeq高通量平台进行2×150 bp的双端测序。采用FastQC(http://www.bioinformatics.babraham.ac.uk/projects/fastqc)对原始数据进行质量控制,使用fastp对数据进行过滤。采用 BWA-MEM 程序将过滤后的高质量数据比对马铃薯参考基因组(DM v6.1),采用GATK软件包来进行SNP的检测及过滤,采用ANNOVAR软件对SNP位点进行注释。
1.3 遗传多样性分析
采用GCTA软件对SNP数据(去除MAF<0.05的SNPs)进行主成分分析。采用fastTree软件中的Maximum likelihood算法构建系统发育树,对系统发育树分支的可靠性进行验证(bootstrap,1 000 replications)。采用 Admixture软件对分型获得的SNP信息进行群体遗传结构分析,设置K=2—20(即假设存在2—20个群体),计算cross-validation(cv)error值,模型选择为混合模型,其余参数为软件的默认设置。根据不同K的cv值选择群体个数。采用stacks程序包中的populations命令进行群体遗传多样性参数的计算。
1.4 选择性消除分析
采用 vcftools程序进行选择性消除参数的计算。利用代表不同田间晚疫病抗性群体的SNP标记信息,以20 kb为窗口,5 kb为步长分别计算π值,并计算代表不同窗口和步长抗病种质和感病种质 π值的比值。同时,选取相同长度的窗口和步长计算不同晚疫病抗性群体间的Fst值。最终获得不同窗口和步长的遗传区段的π值比值和Fst值。其中,抗病和感病种质之间π值比值小,代表相对于感病种质,抗病种质的这段染色体区域核苷酸多样性低,是受选择的遗传区段,选取π值比值最小的5%的遗传区段,并选取这些遗传区段中Fst值最大10%的遗传区段进行后续分析,即选取占基因组0.5%的遗传区段。通过基因本体论分析(gene ontology,GO)预测遗传区段内的基因。基因的氨基酸序列利用 Clustal Omega进行多序列比对。进化树利用MEGA6软件的Neighbor-Joining参数通过计算1 000次生成树图。
1.5 马铃薯抗晚疫病种质全基因组关联分析
以101个抗病种质和21个感病种质的群体SNP信息,利用GEMMA 0.98.1软件的一般线性模型进行全基因组关联分析,得到各SNP位点与性状关联程度的值,并计算-log(),-log()值越大,表示 SNP位点与性状的关联程度越强。利用 CMplot绘制-log()和期望-log()的QQ图以及-log()的曼哈顿图。选择-log()>6的SNP位点周围50 kb的基因组序列,通过基因本体论分析预测遗传区段内的基因。
2 结果
2.1 马铃薯田间晚疫病抗性鉴定
将收集的马铃薯品种(系)经2018—2021年在贵州省毕节市威宁县和七星关区内6个地点种植,进行田间晚疫病抗性鉴定。有101个品种(系),在晚疫病爆发一个月后依然能够存活,基本可以满足当地主栽的中晚熟马铃薯品种生长期90—100 d的要求,确定为抗晚疫病品种。同时以在晚疫病爆发后10 d左右大部分植株死亡为标准,共鉴定得到感病品种175个,进一步从中挑选了薯条/薯片加工品种11个和优秀亲本9个(两者间有重叠)及拥有其他优良性状的品种4个,共21个品种进行下一步分析(表1)。
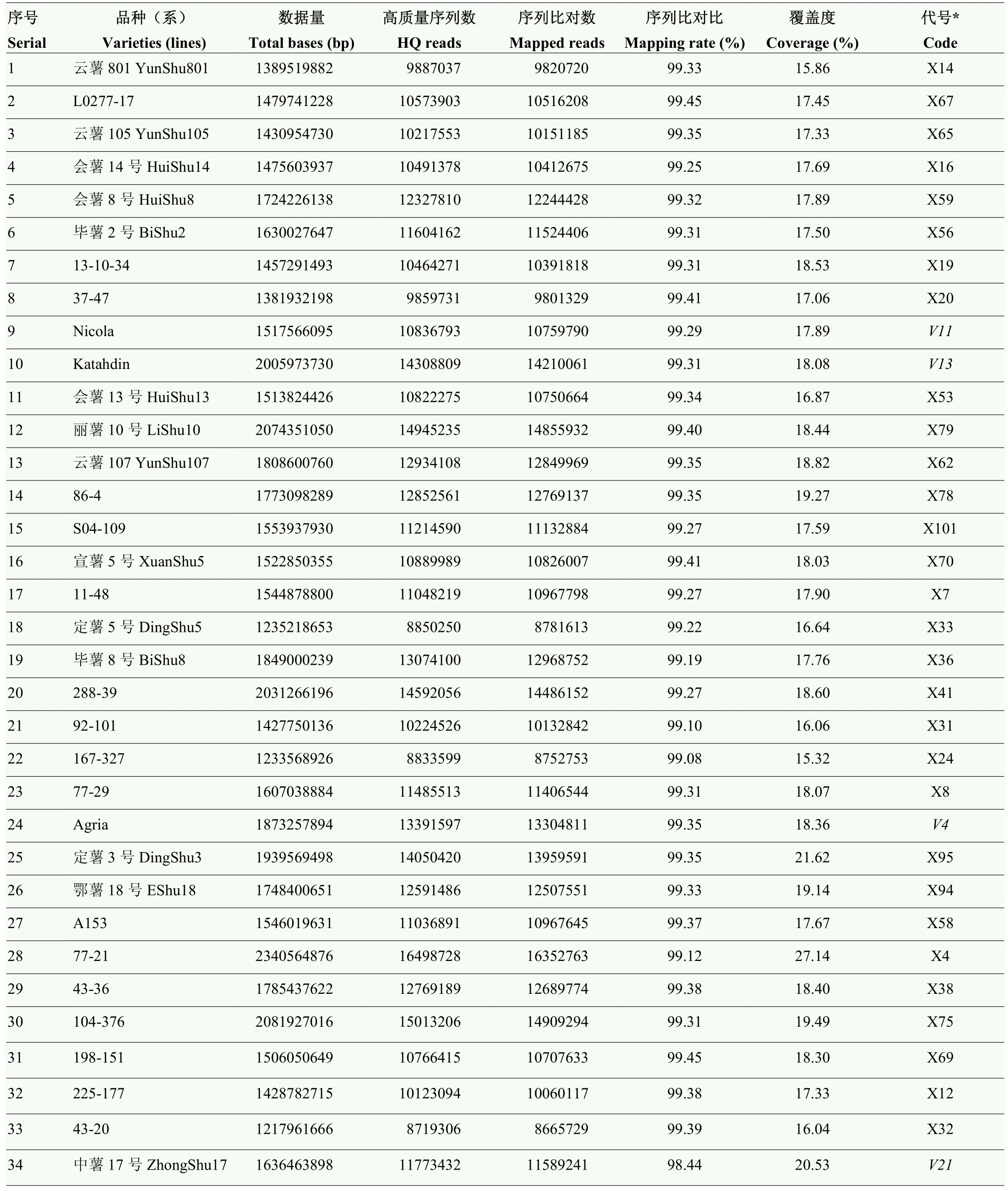
表1 122份马铃薯种质资源和2d-RAD简化基因组测序情况Table 1 The 122 potato germplasm and 2d-RAD simplified genome sequencing results
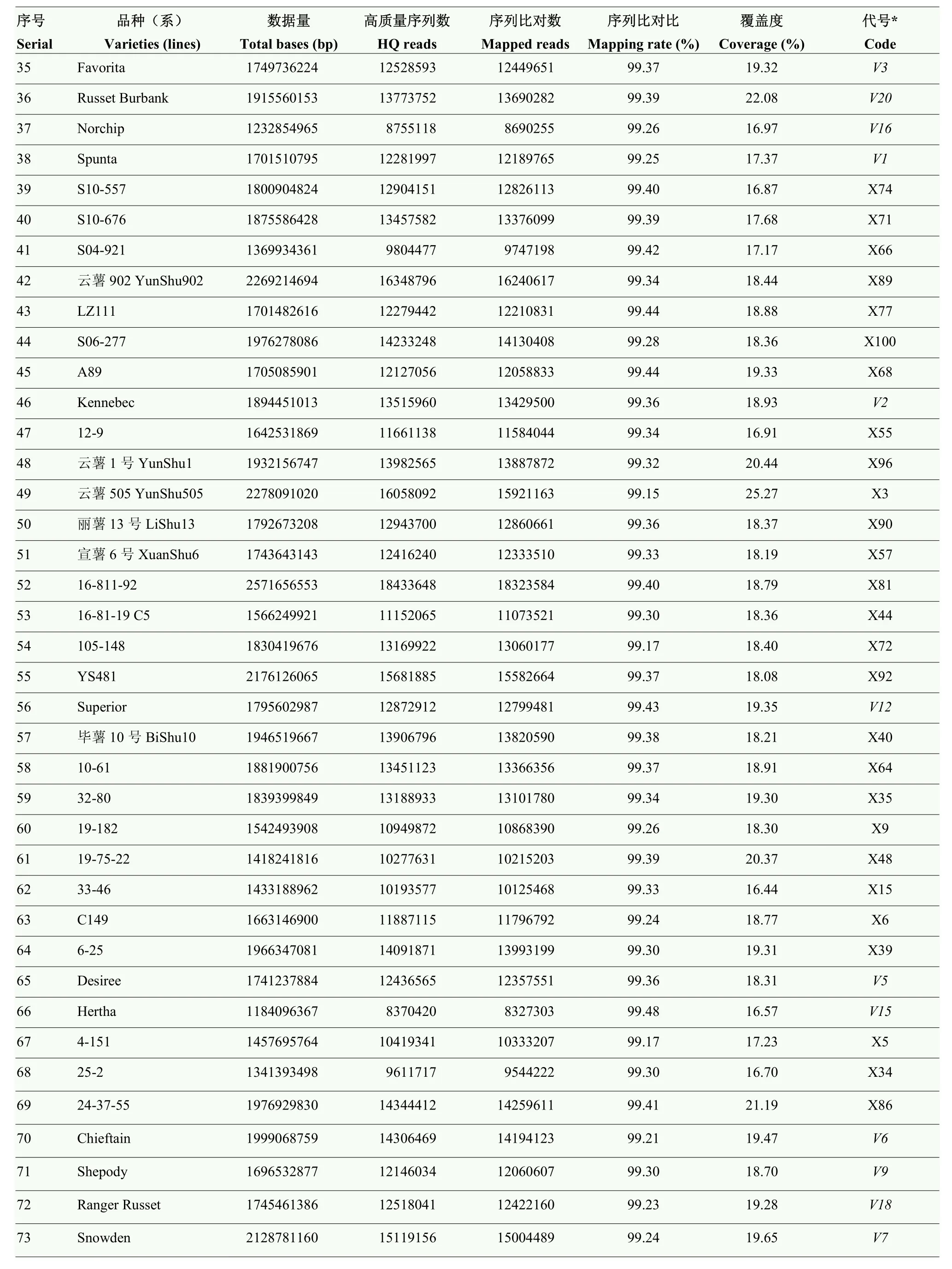
续表1 Continued table 1
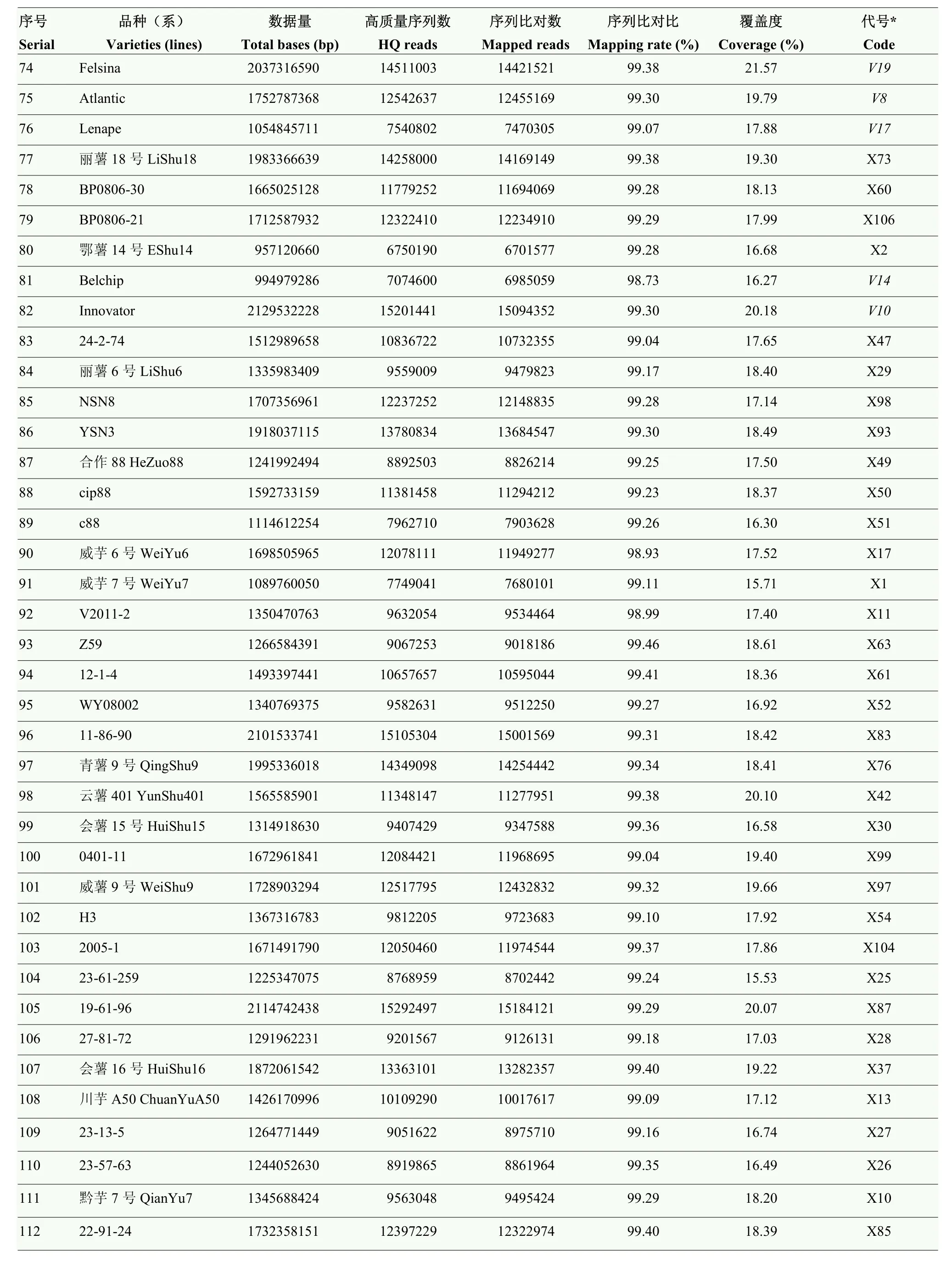
续表1 Continued table 1
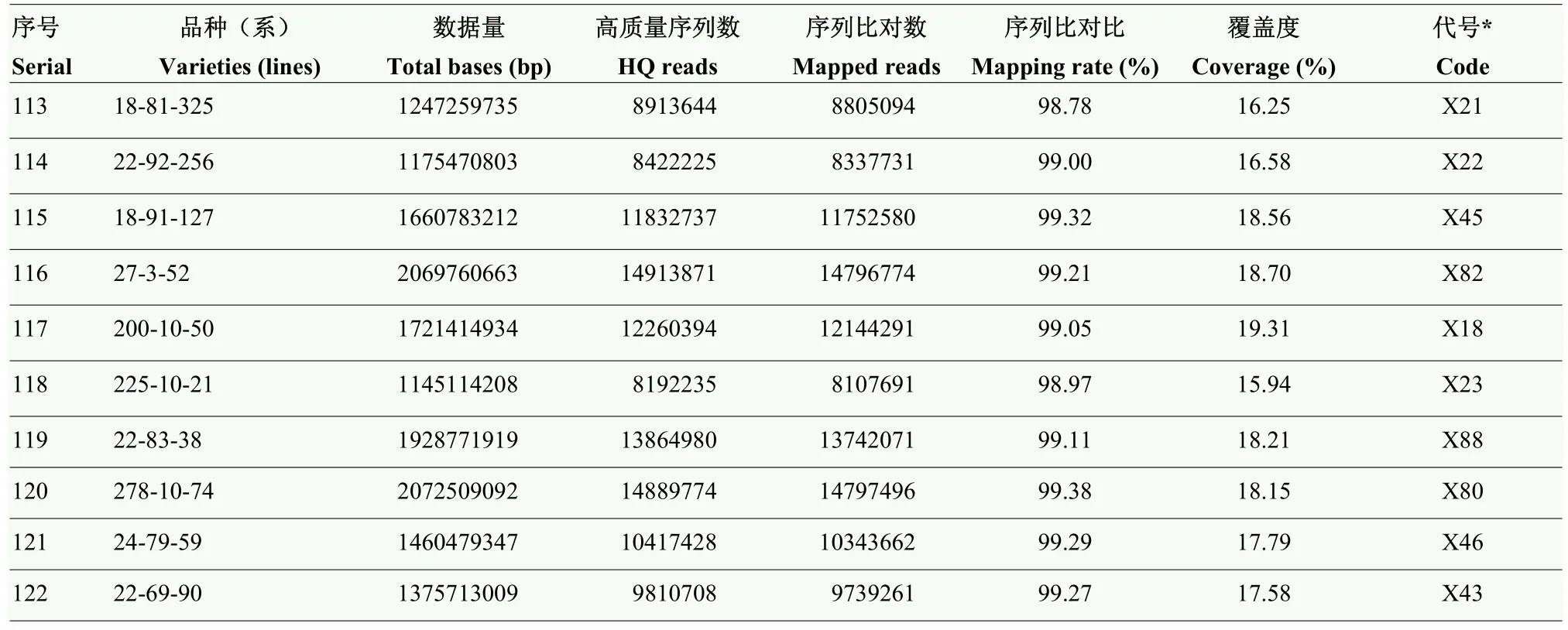
续表1 Continued table 1
2.2 马铃薯种质的简化基因组测序
将122个(101个抗晚疫病马铃薯品种(系)以及21个感病品种)样本进行dd-RAD简化基因组测序。每个样本各产生957 120 660—2 571 656 553 bp的测序数据。过滤掉低质量测序片段(Reads)和不合格序列后,每个样本各产生6 750 190—18 433 648个高质量测序片段,其中,6 701 577—18 323 584个测序片段与参考基因组匹配(DM v6.1),占全部测序片段的99.28%—99.48%,覆盖参考基因组15.32%—27.14%(表1)。共检测到8 697 602个SNP,其中,在基因外显子区域的有397 244个SNP,内含子区域的有970 743个SNP,基因上下游1 kb内有499 056个SNP,基因间区域有6 650 230个SNP(表2)。马铃薯12条染色体上,平均每1 Mb得到的SNP在2 828个以上(图1)。
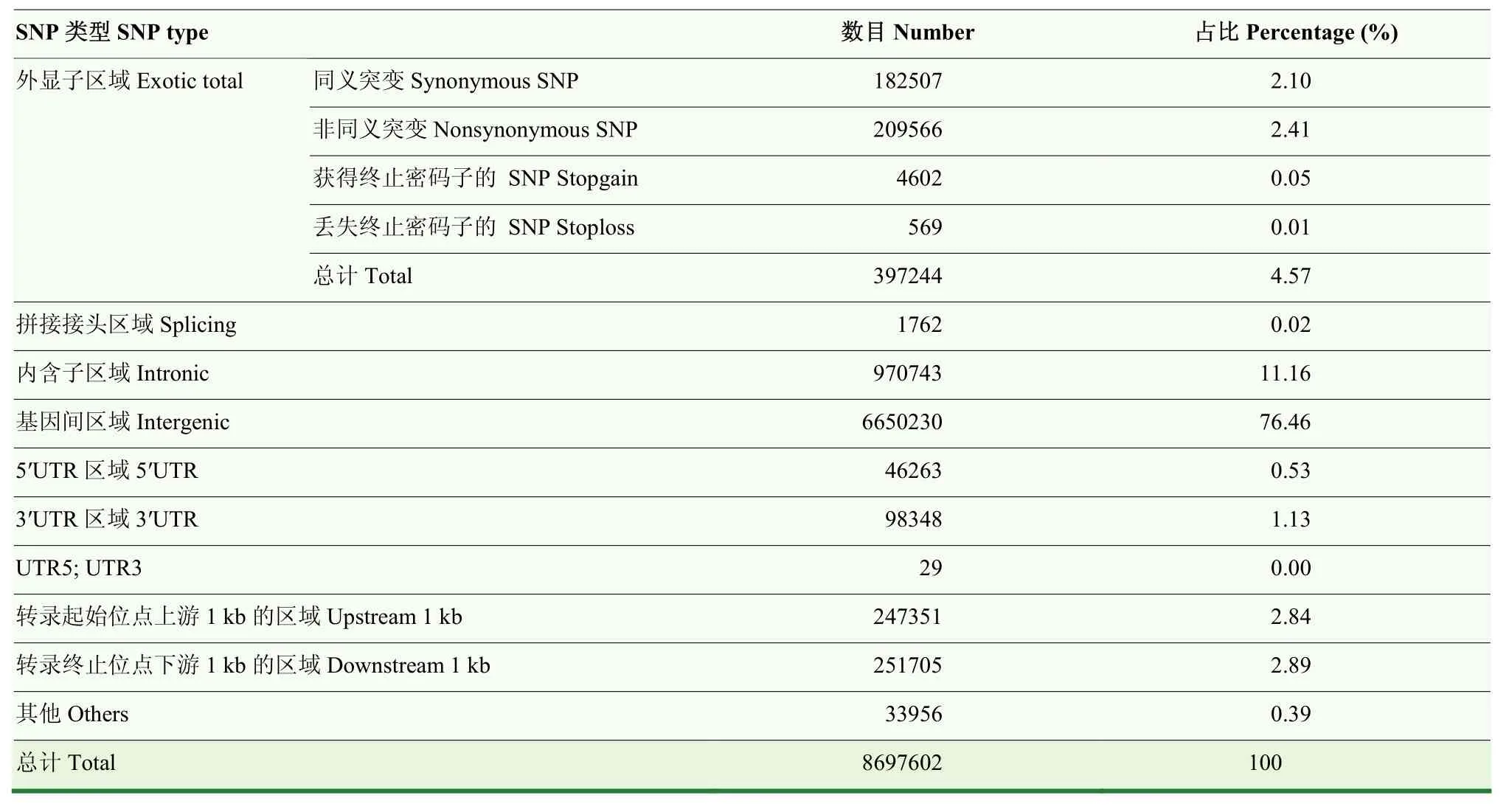
表2 122份马铃薯种质资源SNP基因分型情况Table 2 SNP identification in the 122 potato germplasm
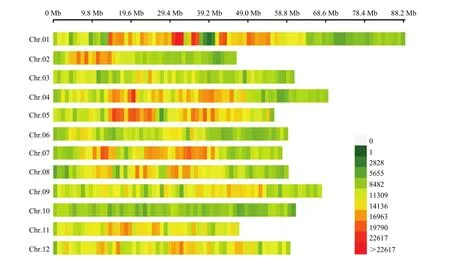
图1 2d-RAD简化基因组测序分型得到的SNP密度热图Fig. 1 SNP density heat map from 2d-RAD simplified genome sequencing
2.3 马铃薯种质的遗传多样性分析
利用检测到的SNP信息对122份马铃薯种质进行种群分析。首先,假定这些种质资源由2—20个群体(population)组成(即K=2—20),利用Admixture软件分析种群结构,当K=6时,cross-validation(cv)error值最小,但当K=3—9时,cv值差别不大(图2-A)。说明这些种质资源可能是由3—9个群体组成,但最可能由6个群体(population)组成(图2-B)。同时,将群体的SNP信息简化为3个维度的信息,进行主成分分析(PCA),将结果根据种群结构分析得到的 6个群体,绘制三维散点图(图2-C),显示虽然各群体成员有聚集在一起的趋势,但界限并不明显。进一步绘制遗传进化树(图2-D),显示这些种质资源可以分成6个群体(population),群体内又有亲缘关系更近的资源聚集成亚群(sub-population),如,在群体Ⅱ中来源于云薯的品种(系)聚集在亚群3中,几个著名的加工品种(如Atlantic、Shepody等)聚集在群体Ⅳ中,亲本来源于国际马铃薯中心的几个品系聚集在群体Ⅵ内的亚群2和亚群3中。同时,发现种群结构的划分与资源的晚疫病抗性并不相关。
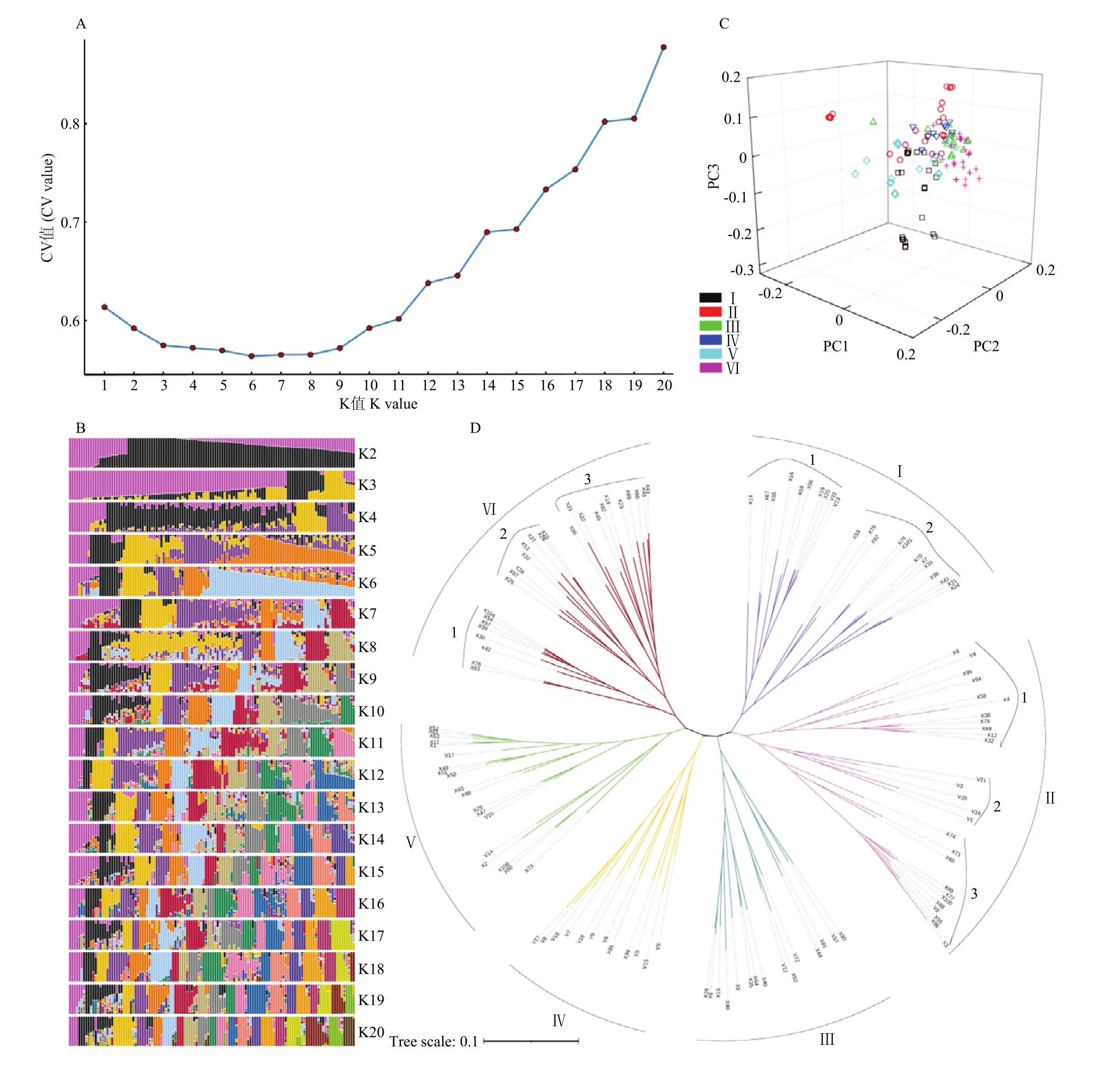
图2 122份马铃薯种质资源的种群分析Fig. 2 Population structure analysis of the 122 potato germplasm
进一步利用种群结构信息进行种质遗传多样性分析。在6个群体内平均核苷酸多样性值(π),范围为0.2055—0.2572(表3),说明群体内样本间的遗传差异较大,范围为0.156909—0.187336(表4),说明群体间遗传分化程度也较大。6个群体内的期望杂合度(e)范围为0.187—0.2297,观测杂合度(o)范围为0.0829—0.1186(表3),观测杂合度均小于期望杂合度。同时6个群体内的近交系数(Fis)范围为0.2412—0.3554(表3),说明6个群体内存在近交的现象。通过进行遗传多样性参数的计算,发现尽管101份马铃薯抗晚疫病种质和21份感病种质存在较丰富单核苷酸遗传多样性,但在选育这些种质资源的过程中存在着亲缘关系较近亲本相互杂交的情况。
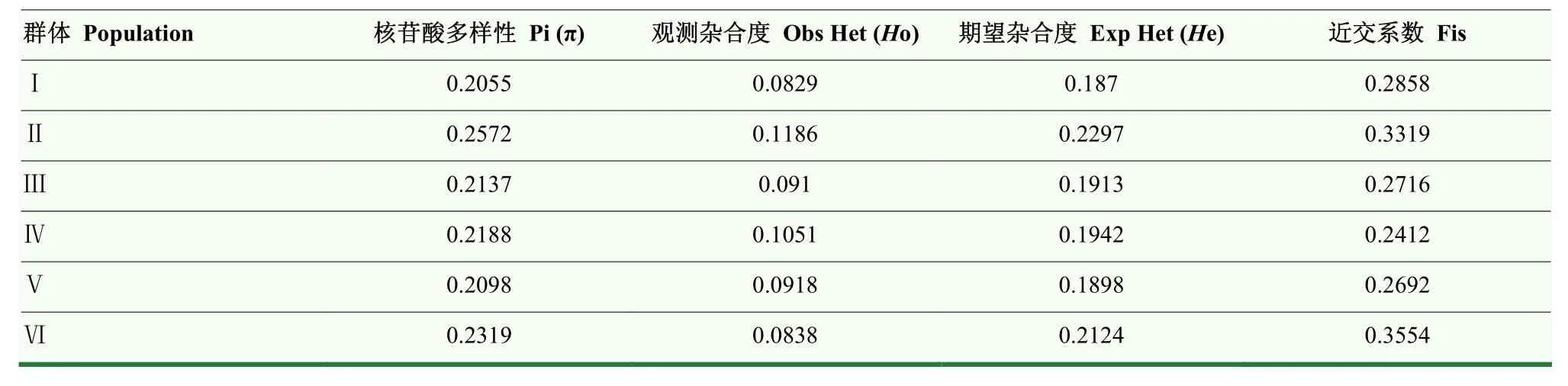
表3 群体内的遗传多样性参数Table 3 Genetic diversity parameters within the population
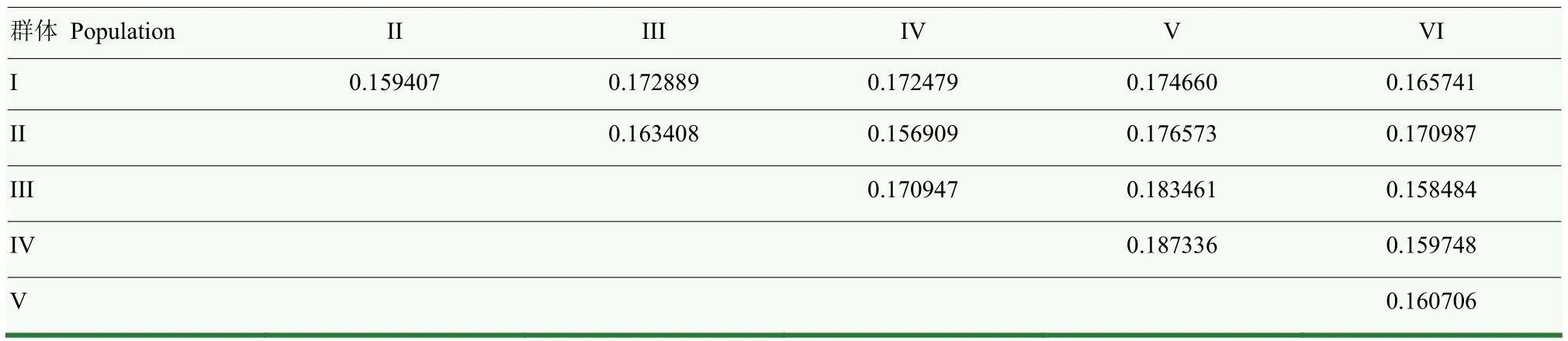
表4 群体间群体分化指数(Fst)的矩阵Table 4 The matrix of fixation index (Fst) among populations
2.4 马铃薯种质选择性消除分析
以分离基因组内可能影响马铃薯晚疫病抗病性状的遗传区段进行选择性消除分析。选取抗病和感病种质之间 π值比值最小的 5%的遗传区段,并进一步选取这些遗传区段中Fst值最大的10%的遗传区段进行后续分析,共获得993个遗传区段,合并相邻的遗传区段后,共有745个遗传区段,其中,最大的遗传区段长75 kb(图3-A)。这些遗传区段中共包含507个基因,通过GO基因功能预测,膜的组成部分有最多数目的基因;与防御反应相关的有14个基因;许多基因预测拥有分子结合功能,如:ATP结合、ADP结合、DNA结合、氨基酸结合、锌离子结合、铁离子结合等(图3-B)。在这些基因中,还发现了 4个拥有晚疫病抗病(R)基因结构域特性的NBS-LRR类基因(图3-C),与已知的晚疫病抗病基因进行序列比对,发现与、的序列相近;、和与/、和的序列相近。
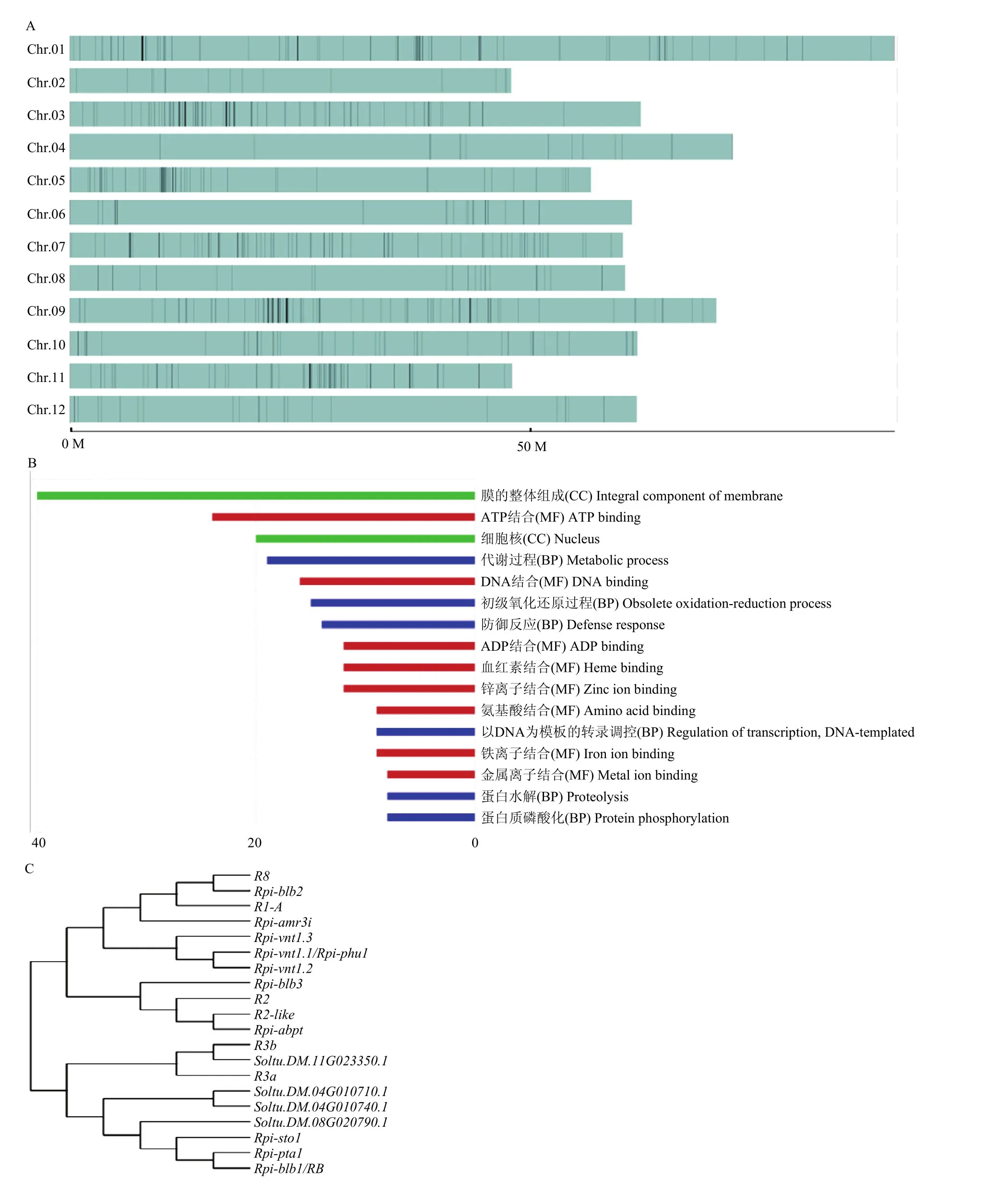
图3 选择性清除分析Fig. 3 Selective sweep analysis
2.5 马铃薯种质全基因组关联分析
以101个抗病种质和21个感病种质的群体SNP信息,进行全基因组关联分析,得到各SNP位点与性状关联程度的-log()。在QQ图右侧,部分SNP位点的观测-log()与期望-log()发生偏离(图4-B),说明与总体SNP位点不同,这部分SNP的观测-log()不符合正态分布,表明这部分SNP位点附近的染色体区域可能受到了选择。在曼哈顿图中,与晚疫病抗性性状关联最强的9个SNP位点(-log()>6)分布于马铃薯的不同染色体上(图4-A)。在这些SNP位点周围50 kb的基因组范围内,共有69个基因,其中,41个基因可以依据细胞成分、生物过程及分子功能通过GO预测得到基因功能。在41个基因中,12个基因编码的蛋白预测为质体的组成成分,15个基因预测参与到应激反应,12个基因预测参与清除过氧化物自由基的过程,4个基因预测参与半胱氨酸合成,3个基因预测参与跨膜运输(图4-C)。
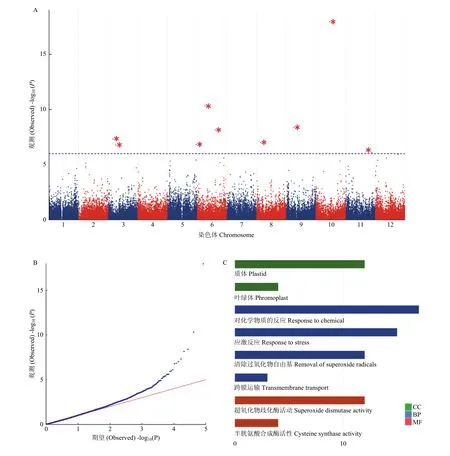
图4 全基因组关联分析Fig. 4 Genome-wide association analysis
3 讨论
3.1 dd-RAD简化基因组测序是马铃薯基因分型的有效手段
构建dd-RAD简化基因组文库首先通过对参考基因组进行分析,选取合适的限制性内切酶对基因组DNA进行酶切,因此,在样本间基因组DNA酶切位置基本相同,并且可以在基因组内获得位置间隔相对均匀的待测序片段。相比较于一些利用物理手段进行基因组 DNA片段化的文库构建方法,dd-RAD是一种可预测的文库构建方法。文库构建完成后,通过2×150 bp的双端测序,得到150 bp的测序片段,相对于其他利用限制性内切酶构建测序文库的方法,如:2b-RAD只得到36 bp的测序片段,dd-RAD得到的测序片段长度更长,因此,将测序片段比对到参考基因组上时,比对结果更加可靠,分型结果也更加准确。本研究利用 dd-RAD简化基因组测序,对122份马铃薯种质资源进行基因分型,测序片段覆盖了参考基因组 15.32%—27.14%,共检测到8 697 602个SNP标记。在马铃薯12条染色体上,平均每1 Mb含有2 828个SNP以上(图1)。马铃薯的连锁不平衡(LD)衰减值最小为600 kb,平均在2—4 Mb,表明本研究的SNP密度足以满足要求。
3.2 群体结构分析揭示马铃薯种质资源之间的遗传关系
群体结构分析能够揭示马铃薯种质资源之间的遗传关系。研究表明,通过群体结构分析,可以将普通栽培种、安第斯栽培种及野生种区分开。并且,部分来源相同、育种地相近的种质,可以在群体结构分析中聚集在一起。如,DUAN等研究表明大部分中薯系列品种聚集在一个类群中。WANG等研究表明不同时间引入中国的国外品种、来源于国际马铃薯中心的种质资源、以及中国不同地区选育的马铃薯品种分别聚集在不同的亚群。与这些研究类似,本研究来源于云薯的种质聚集在群体Ⅱ的亚群3中,亲本来源于国际马铃薯中心的几个品系聚集在群体Ⅵ的亚群 2和亚群3中(图2-D)。
群体结构分析可以在一定程度上区分薯条、薯片加工品种与其他普通栽培种,本研究几个著名的加工品种(如大西洋(Atlantic)、夏坡蒂(Shepody)等)聚集在群体Ⅳ中(图2-D)。但种质种群结构的划分与种质的晚疫病抗性并不相关(图2-D),这可能是由于晚疫病抗性基因在群体里属于稀有位点,它们的选择与否与群体结构之间没有必然联系。
同时,为了缓解马铃薯遗传基础狭窄的问题,可以根据群体结构,分析种质资源之间的亲缘关系,并在杂交育种中选择亲本亲缘关系较远的种质。例如,本研究所涉及的几个易感晚疫病品种Agria、Desiree、Innovator、Felsina、Nicola和Superior,它们除了拥有优良的农艺性状外,更重要的是它们具有较高的一般配合力,是国际范围内近期作为直接亲本育成品种数最多的几个品种,可以在亲本选配中选择群体Ⅵ内的种质杂交。同时,本研究中所涉及的种质,尤其是感病种质较少,可以利用在不同研究中相同种质所处的种群结构位置,分析其他研究中的种质与本研究种质间的亲缘关系。如通过相同品种(Favorite、Desiree、中薯17号)分析与189份不同晚疫病抗性的普通栽培种、安第斯栽培种及野生种之间的亲缘关系。通过相同的云薯系列品种,分析与292份不同时间引入中国的国外品种、来源于国际马铃薯中心的种质资源、以及中国不同地区选育的马铃薯品种之间的关系。通过相同品种(Atlantic、Favorita、Katahdin)分析与209份中国育成的品种之间的亲缘关系。
3.3 马铃薯田间抗晚疫病种质资源拥有较丰富的单核苷酸多态性但在其选育过程中存在近交现象
马铃薯作为营养器官繁殖的作物,其品种选育经历的有性杂交过程十分有限。系谱学研究显示,有记录的马铃薯杂交育种始于一个智利普通栽培品种Rough Purple Chili,它的后代与欧洲品种杂交后,表现突出,从此马铃薯杂交育种开始得到重视并广泛应用。美国马铃薯协会列出北美177个应用于生产的品种,其中,156个品种含有Rough Purple Chili的血缘。在全世界育成的有记录的4 397个品种中,15个直接亲本育成的品种数占总品种数的 14.9%;523个有记录的中国品种中,8个直接亲本育成的品种数占总品种数的27.34%。因此,马铃薯存在着种质资源的遗传基础狭窄的问题。在本研究中,通过利用122份马铃薯种质的群体 SNP信息,可以将这些种质资源分成6个群体(图2)。6个群体内的观测杂合度均小于期望杂合度,并且 6个群体内的近交系数(Fis)为 0.2412—0.3554(表3),说明选育这些种质的过程中存在近交的现象,也反映了这些种质资源的遗传基础狭窄的问题。同时,马铃薯作为基因组高度杂合的多倍体,本身拥有较丰富的单核苷酸多态性。在6个群体内,反映群体内样本间平均遗传多样性差异程度的平均核苷酸多样性值(π)为0.2055—0.2572(表3),反映群体间的分化程度群体分化指数(Fst)为 0.156909—0.187336(表4),说明田间抗晚疫病种质资源拥有较丰富的单核苷酸多态性。
3.4 选择性清除分析和关联分析有助于分离影响农艺性状染色体区段
选择性清除分析利用在受选择的遗传区段中,遗传多样性降低并且遗传分化升高,来分离影响农艺性状的染色体区段。本研究以20 kb为窗口5 kb为步长,在基因组相同位置,分别计算不同晚疫病抗性马铃薯种质间平均核苷酸多样性(π)值比值和群体分化指数(Fst)值。相较于其他类似的研究,本研究选择了相对较小的窗口计算π值和Fst值,一方面是因为简化基因组测序只对部分基因组进行了基因分型,过大的窗口可能平均化已测序和未测序遗传区段的单核苷酸多态性信息,并导致结果不能反映该区段的真实情况;另一方面本研究种群结构的划分与其晚疫病抗性并不相关,选择较小的窗口更能精细化区分遗传区段的受选择情况。以 π值比值极小及Fst值极大,选择可能影响晚疫病抗性的745个遗传区段进一步分析,这些遗传区段中共包含507个基因,其中,有14个基因可能参与防御反应,更为重要的是有4个类基因,1个与的序列相近,另外3个与拥有广谱晚疫病抗性、和的序列相近(图3-C),其中,/克隆自马铃薯野生种,是克隆的第一个具有广谱晚疫病抗性的基因,和克隆自马铃薯野生种,是的同源基因。说明选择性清除分析有助于分离影响晚疫病性状的遗传区段。
关联分离利用连锁不平衡,使用自然群体挖掘与性状相关联的基因,在马铃薯中也有广泛的应用。例如,通过分析抗黄瘟病()基因的等位基因座,在双倍体及四倍体马铃薯群体中,对抗黄瘟病性状与SNP标记之间进行了关联分析,克隆出了一个抗黄瘟病的基因。同样也有研究利用全基因组关联分析对马铃薯产量、淀粉含量、抗旱等性状进行了研究。在本研究中,得到了 9个可能与晚疫病性状相关联的SNP位点,在它们周围的50 kb的基因组范围内,有15个基因预测参与应激反应,有12个基因预测参与清除过氧化物自由基,这些基因可能参与了晚疫病侵染后的细胞的修复过程(图4-C)。
4 结论
dd-RAD简化基因组测序可以在马铃薯基因组中分型到数量较多分布相对均匀的SNP标记。马铃薯田间抗晚疫病种质资源拥有较丰富的单核苷酸多态性但在其选育过程中存在近交的现象。选择性清除和关联分析有助于分离影响晚疫病抗病性状的遗传区段。
