Pressure-induced phase transition in transition metal trifluorides
2022-10-26PengLiu刘鹏MeilingXu徐美玲JianLv吕健PengyueGao高朋越ChengxiHuang黄呈熙YinweiLi李印威JianyunWang王建云YanchaoWang王彦超andMiZhou周密
Peng Liu(刘鹏) Meiling Xu(徐美玲) Jian Lv(吕健) Pengyue Gao(高朋越) Chengxi Huang(黄呈熙)Yinwei Li(李印威) Jianyun Wang(王建云) Yanchao Wang(王彦超) and Mi Zhou(周密)
1State Key Laboratory of Superhard Materials&International Center for Computational Method and Software,College of Physics,Jilin University,Changchun 130012,China
2Laboratory of Quantum Functional Materials Design and Application,School of Physics and Electronic Engineering,Jiangsu Normal University,Xuzhou 221116,China
3MIIT Key Laboratory of Semiconductor Microstructure and Quantum Sensing,Nanjing University of Science and Technology,Nanjing 210094,China
Keywords: high-pressure structure transition,crystal structure prediction,high-pressure x-ray diffraction experiments,transition metal
1. Introduction
Transition metal trifluorides,a class of materials with the chemical formulaMF3(M=Sc, Ti, V,Cr, Mn, and so forth)have attracted considerable attentions owing to their versatile applications in negative thermal expansion materials,[1–4]batteries,[5–7]and hydrogen storage materials.[8–12]More importantly, they have been demonstrated to be ideal materials to study the Jahn–Teller and spin–orbit coupling effects.[13,14]Under ambient conditions,MF3usually adopts a simple perovskite-like structure with a completely vacant A site,[15–19]in which the metal atom is surrounded by a tilted octahedron of corner-shared fluorine atoms. Moreover, the tilting angle of the octahedron decreases with increasing temperature, causing a high-temperature phase transition to the cubic ReO3-type structure.[17,20]It is essential to note that the physical and chemical properties ofMF3are generally associated with structural parameters, such as polyhedral volume and octahedral tilt.[21–28]Therefore, investigating the structural changes inMF3will provide new insights for designing functional materials.
It is well known that pressure is a key thermodynamic variable that modifies the crystal structure and effectively controls material properties. For example, high-pressure experiments have led to the discovery of novel materials with unique properties (e.g., high-temperature superconductors such as H3S,[29,30]LaH10,[31,32]and C–S–H[33]). The high pressure thus offers exciting opportunities for discovering new materials that do not exist under ambient conditions.[34–40]Highpressure does not necessitate the destruction of theMF6octahedron inMF3systems. In practice, the pressure-induced structural evolution is only the cooperative tilting of theMF6octahedra,[41–43]which can be summarized as follows: (i) an elongation of theMF6octahedra along thecaxis leads to a small octahedral strain, (ii) theMF6octahedral strain disappears, and(iii)MF6octahedral elongation occurs along theaaxis.
In this work,we adopted a combination of first-principle calculations and experiments to explore the high-pressure phase of TiF3. Our results suggest that TiF3transforms from the rhombohedral (R–3c) phase to an orthorhombic (Pnma)phase at high pressure,accompanied by the destruction of the TiF6octahedra and formation of TiF8square antiprismatic units. The high-pressurePnmaphase of TiF3is confirmed by the laser-heated diamond-anvil-cell experiment and shows semiconducting character with a band gap of 2.65 eV.We further confirmed that the pressure-induced transition fromR–3ctoPnmaphase is a general trend in transition metal trifluorides,such as ScF3,VF3,CrF3,and MnF3.
2. Methods
Ab initiocalculations The search for TiF3structures(1–4 formula units) was performed at pressures of 20 GPa and 50 GPa via an unbiased swarm intelligence based method,Crystal structure AnaLYsis by Particle Swarm Optimization(CALYPSO),[44–46]which is designed to search for the most stable or metastable structures of given compounds.[47–57]Our first-principle calculations were based on density functional theory,[58]as implemented in the VASP package.[59]The core electrons were treated by the projector-augmented wave approximation,[60]and the exchange–correlation functional was given by the generalized gradient approximation parameterized by Perdew, Burke, and Ernzerhof.[61]The planewave cutoff energy was set to 800 eV, and Monkhorst–Packkmeshes with a spacing of 2π×0.03 ˚A-1were chosen for Brillouin zone sampling to ensure that all the energy calculations converged well to~1 meV/atom. The Heyd–Scuseria–Ernzerhof(HSE)hybrid functional[62]was employed to accurately evaluate the electronic properties.The dynamic stability of the predicted structure was verified by phonon dispersion analysis using the direct supercell method,as implemented in the PHONOPY code.[63]
Experimental procedures TiF3was obtained from Alfa Aesar and verified by powder x-ray diffraction (XRD).[16]TiF3powder, together with a ruby ball, was loaded into a symmetric diamond-anvil-cell (DAC) with a culet size of 320 μm with no pressure transmitting medium, and the pressure was determined by ruby fluorescence.[64]The sample was first compressed to 20 GPa and then heated to approximately 2000 K using a laser heating system with a diode-pumped CW ytterbium fiber laser(central wavelength of 1080 nm and maximum power of 100 W).Synchrotron XRD patterns were recorded at beamline BL10XU of Spring-8 (Japan) with a wavelength of 0.414 ˚A,and the refinement was fitted using the GSAS software[65]and EXPGUI interface.[66]In situelectrical conductivity measurements, under high pressure and low temperature, were conducted in a DAC equipped with a van der Pauw-type microcircuit.[67]
3. Results and discussion
TiF3usually adopts a VF3-type structure with a space group ofR–3cat ambient pressure,[16,68]in which Ti is surrounded by a tilted octahedron of corner-shared fluorine atoms. The tilting angle of the octahedra decreases with an increase in temperature,leading to a phase transformation from rhombohedral to cubic at 370 K.[20]The cubic structure with thePm-3mspace group is isostructural in ReO3, consisting of the TiF6octahedra without tilt fluctuations. To determine the high-pressure structure of TiF3, we performed extensive structural searches at pressures of 20 GPa and 50 GPa. In our structural searches, all the experimental structures of the TiF3,R–3c,andPm-3mphases were successfully reproduced using the CALYPSO method, validating the reliability of our structure-searching method. In addition to the known experimental structures, an orthorhombic structure with the space group ofPnmawas successfully observed at 20 GPa.
Enthalpy as a function of pressure for thePnmaphase relative to theR-3cphase is shown in Fig. 1(a). It is apparent that the ambient-pressure phase ofR–3ctransforms to thePnmaphase at 12 GPa, where the F atoms are in square antiprismatic coordination of the Ti atoms (Fig. 1(b)), which is isostructural to YF3at ambient pressure.[69]It is generally accepted that high-pressure phases of lighter elements or compounds in the periodic table are expected to be identical to the ambient structures of the corresponding heavier elements or compounds.[70]At 20 GPa,the largest and average Ti–F bond lengths in thePnmaphase are 2.12 ˚A and 2.05 ˚A,respectively,while all the bond lengths of Ti–F are equal to 1.93 ˚A in theR–3cphase. Furthermore, the coordination number of Ti increases from 6 to 8,weakening individual Ti–F bonds and inducing longer Ti–F bond lengths.Interestingly,compared with theR–3cphase of TiF3,in which the A-cation site of the perovskite structure is unoccupied,the newly found high-pressurePnmaphase of TiF3can be considered a variant perovskite structure with a completely vacant B site. Thus,under certain circumstances,increasing the pressure has demonstrated to be an efficient strategy to tune the vacant coordination sites of cations in perovskites.
We calculated the phonon dispersions of the predictedPnmaphase of TiF3at 20 GPa (Fig. 1(c)) and observed no imaginary frequencies, indicating that the predicted structure is dynamically stable. Our systematic assessment of energetic and dynamic stabilities suggests that thePnmaphase of TiF3could be realized experimentally. To verify our theoretical predictions,we performed high-pressure measurements on TiF3. The synchrotron XRD pattern of TiF3was obtained at 20 GPa after laser heating to approximately 2000 K,with Rietveld fitting as shown in Fig.1(d). The obtained peaks agree well with the predicted orthorhombicPnmastructure. The refined lattice parameters of the orthorhombicPnmastructure area=5.14 ˚A,b=6.25 ˚A, andc=4.38 ˚A, which are in excellent agreement with our theoretical results.
After the successful synthesis of thePnmaphase in TiF3,we investigated its bonding characteristics and electronic properties. To determine the nature of the bonding,we examined the electron localization function. A less localized charge distribution is observed in the Ti–F bonds(Fig.2(a)),indicating a significant degree of ionicity between the F anions and Ti cations. Furthermore, from Bader charge analysis,[71]the charge values on Ti and F were calculated at 20 GPa. There is a charge transfer of 1.89efrom Ti to F,comparable to that of typical ionic compounds of NaCl.[72]Moreover,the electronic band structure calculations at the HSE hybrid functional level demonstrated that thePnmaphase of TiF3is a semiconductor with a band gap of 2.65 eV(Fig.2(b)). To verify the electrical characteristics of the newly foundPnmaTiF3,anin situhigh pressure electrical conductivity measurement was conducted,based on the van der Pauw-type microcircuit technique.[73]As shown in Fig.2(c),the electrical resistance monotonically increases with decreasing temperature,confirming the semiconductor characteristics ofPnmaTiF3.

Fig.1. (a)Enthalpy vs. pressure curves for Pnma phase of TiF3 relative to the R–3c phase. (b)Crystal structure of the Pnma phase formed by TiF8 square antiprismatic units. The Pnma structure contains 16 atoms/cell,wherein Ti atoms occupy the 4c(0.13,0.75,0.47)positions and the F atoms occupy the 8d (0.17, 0.06, 0.65) and 4c (0.03, 0.25, 0.13) positions. At 20 GPa, the optimized structural parameters are a=5.30 ˚A, b=6.24 ˚A,and c=4.40 ˚A.(c)Phonon dispersion relations of the Pnma phase at 20 GPa. Here,the fractional coordinates of high-symmetry k points are given as follows:Γ(0,0,0),X(1/2,0,0),Y(0,1/2,0),Z(0,0,1/2),R(1/2,1/2,1/2),S(1/2,1/2,0),T(0,1/2,1/2),U(1/2,0,1/2). (d)Measured powder x-ray diffraction(XRD)pattern of TiF3 at 20 GPa with Le Bail method(XRD 2D image is given on the top).Vertical ticks correspond to Bragg peaks of the Pnma structure(pink). The refined lattice parameters of the orthorhombic Pnma structure from the XRD data are a=5.14 ˚A,b=6.25 ˚A,and c=4.38 ˚A.The x-ray wavelength is 0.414 ˚A.
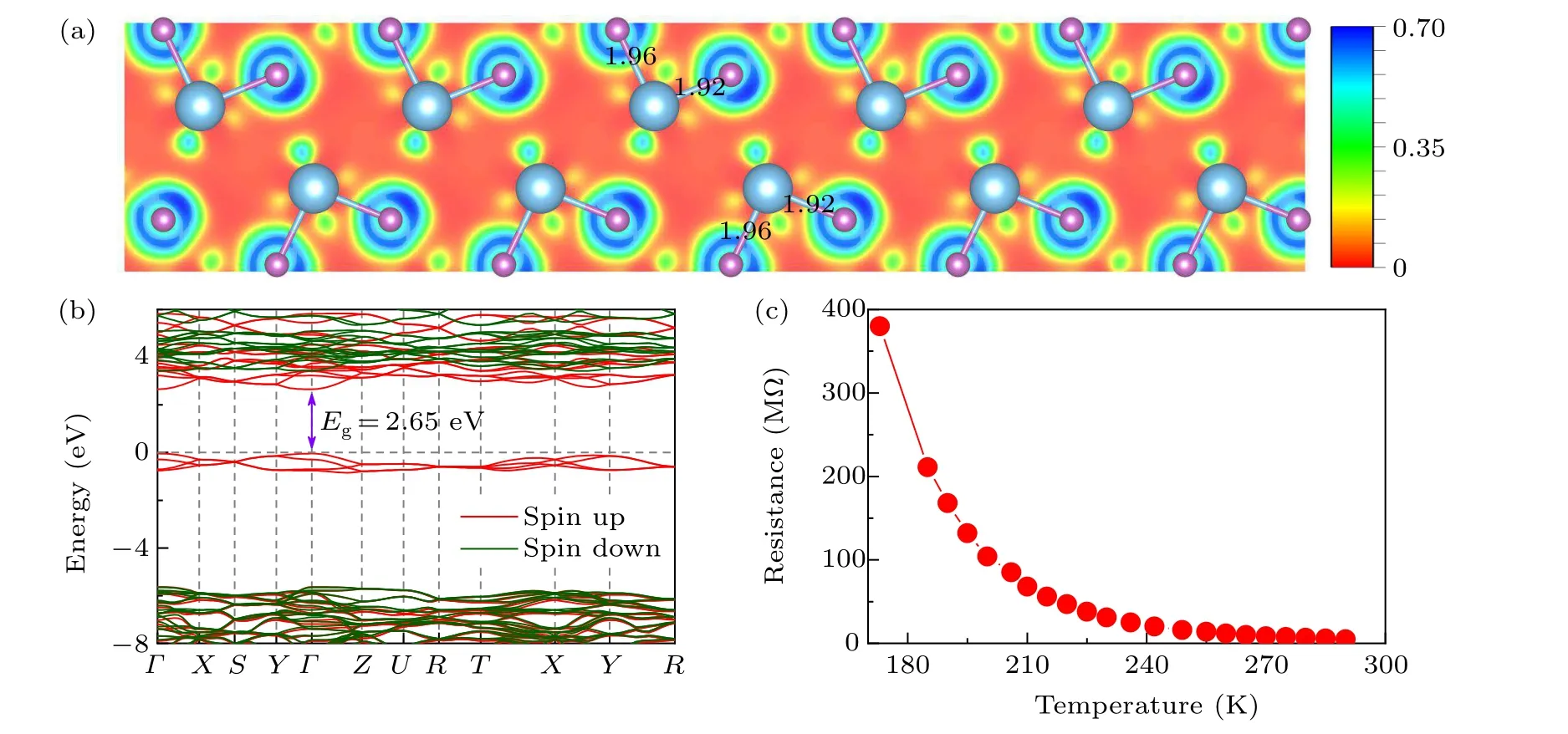
Fig. 2. (a) Calculated ELF of the Pnma phase on the (0 1 0) plane at 20 GPa, in which the bond lengths (in units of ˚A) of the Ti–F bonds are shown. (b) Band structures of the Pnma structures at 20 GPa. The red and green colors denote the spin-up and spin-down bands, respectively.The energy of the topmost valence band state is set to 0 eV.Here,the high-symmetry k points are the same as those in Fig.1(c). (c)Experimental resistance–temperature curve of TiF3 at 20 GPa.
Considering that theR–3cphase is a prototype structure of transition metal trifluorides under ambient conditions and the discovery of the high-pressure phase ofPnmain TiF3,we deliberated whether this pressure-induced phase transition is a common phenomenon in transition metal trifluorides. Thus,the neighboring metal elements Sc, V, Cr, and Mn were chosen. The enthalpies of thePmnaphase as a function of pressure with respect to theR-3cphase for ScF3, VF3, CrF3, and MnF3were calculated and shown in Fig. 3. The phase transition fromR–3ctoPnmais a general trend in those metal trifluorides,and the corresponding pressures are calculated to be 5 GPa,33 GPa,112 GPa,and 40 GPa for ScF3,VF3,CrF3,and MnF3,respectively. Therefore,the predictedPnmaphase could be a prototype structure widely adopted by transition metal trifluorides at high pressure.
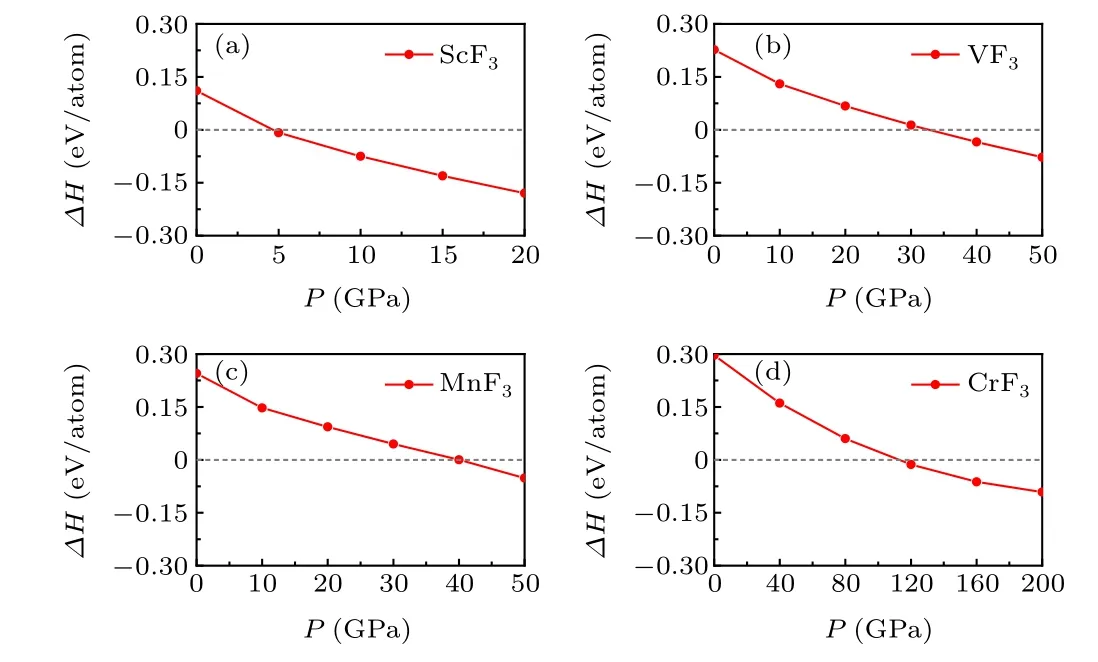
Fig. 3. Calculated enthalpies of Pmna phase as functions of pressure with respect to R–3c phase for ScF3(a),VF3(b),MnF3(c),and CrF3(d)systems.
4. Conclusion
In summary, by combining structure-searching methods with first-principle calculations, a pressure-inducedR-3ctoPnmaphase transition was predicted in TiF3, which was further confirmed by high-pressure experimental synthesis.The first-principle calculations and electrical measurements demonstrated that the high-pressurePnmaphase of TiF3exhibits semiconducting characteristics. Further,since theR–3cphase of TiF3is a prototype structure for transition metal trifluorides at ambient pressure, it was shown that the pressureinduced phase transition fromR-3ctoPnmais a general trend in transition metal trifluorides.
Acknowledgments
Project supported by the National Natural Science Foundation of China (Grant Nos. 12034009, 91961204, and 11974134). Part of the calculation was performed in the high-performance computing center of Jilin University and the School of Physics and Electronic Engineering of Jiangsu Normal University.
猜你喜欢
杂志排行

Chinese Physics B的其它文章
- Design of vertical diamond Schottky barrier diode with junction terminal extension structure by using the n-Ga2O3/p-diamond heterojunction
- Multiple modes of perpendicular magnetization switching scheme in single spin–orbit torque device
- Evolution of the high-field-side radiation belts during the neon seeding plasma discharge in EAST tokamak
- Phase-matched second-harmonic generation in hybrid polymer-LN waveguides
- Circular dichroism spectra of α-lactose molecular measured by terahertz time-domain spectroscopy
- Recombination-induced voltage-dependent photocurrent collection loss in CdTe thin film solar cell