Genetic Differentiation Among Populations of Octopus minor Based on Simple Sequence Repeats Mined from Transcriptome Data
2022-10-24NANZeXURanHOUChunqiangandZHENGXiaodong
NAN Ze, XU Ran, HOU Chunqiang, and ZHENG Xiaodong, *
Genetic Differentiation Among Populations ofBased on Simple Sequence Repeats Mined from Transcriptome Data
NAN Ze1), 2), XU Ran1), 2), HOU Chunqiang3), and ZHENG Xiaodong1), 2), *
1)Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao 266003, China 2) Key Laboratory of Mariculture, Ocean University of China, Qingdao 266003, China 3) Tianjin Fisheries Research Institute, Tianjin 300457, China
(Sasaki 1920) is an important commercial cephalopod species in China. This species has declined sharp- ly in recent years. Hence, genetic studies ofare imperative to exploit and manage the wild resource. In this study, 46192 microsatellite loci were discovered in 70174 unigenes from the transcriptomic data. Among all of the simple sequence repeat (SSR) unit categories, di-nucleotide and tri-nucleotide SSRs accounted for 45.26% and 14.49%, respectively. A total of 108 SSRs were test- ed, of which 21 were neutral and polymorphic. Seven SSRs were selected for genetics analyses of thepopulations in the Bohai Sea, the Yellow Sea, and the southwest coast of the Taiwan Strait region. Significant pairwisestvalues were detected among the samples. The UPGMA tree based on genetic distances suggested that the sampling locations could be arranged in three clusters. These markers are evidence that the populations in this region may be structured, with samples from Penghu differing remarkably from those in northern China. The present study characterized genetic markers for population assessment, management, and conser- vation of.
; transcriptome; simple sequence repeats; genetic divergence
1 Introduction
(Sasaki 1920) is widely distributed along the coastal waters of China, the Korean Peninsula, and the Japanese archipelago (Yamamoto, 1942; Kim., 2008). It is an important commercial fishery in China, Korea, andJapan. Many studies ofin physicology (Seol., 2007; Chen., 2019), aquaculture (Song., 2019), and genetics (Wang., 2017) have been reported. How-ever, overfishing has led to a sharp decline in the abun- dance of this species in recent years. Thus, it is crucial to study the genetic diversity and population structure for ra- tional utilization of.
Simple sequence repeats (SSRs), or microsatellites aresimple tandem repeat DNA sequences consisting of 1–6 basesthat are widely distributed in the genome. Because of high polymorphism, neutrality, and codominance, SSRs have become an effective tool to investigate population genetics. Microsatellite markers inhave been de- veloped using the magnetic bead enrichment method (Zuo., 2011), and population genetics analyses have been carried out using the markers developed with this method (Kang., 2012; Gao., 2016). However, no study has characterized microsatellites based ontrans- criptome data. A large number of SSRs have been iden- tified by next-generation sequencing, providing numerous molecular markers to assess population diversity and ge- netic structure, which will contribute to the conservation of this species.
In the last two decades, the development of molecular tools has contributed greatly to genetic studies (Moreira., 2011; Xu., 2018) and the identification of cryp- tic species (Allcock., 2015; Barco., 2016; Tang., 2020). Many studies have reported the genetic diver- sity and structure ofusing morphological diag- nostic (Gao., 2019) and molecular methods, such as mitochondrial DNA (Sun., 2010; Xu., 2018) and microsatellite markers (Kang., 2012; Gao., 2016). These studies have provided valuable information about the population genetic diversity and structure ofin Chinese waters; however, little is known about the ge- netic divergence between the Chinese populations (parti- cularly the South China Sea population) and other East Asian populations. The Korean Peninsula is bordered by China, so comparing the population genetics between Ko- rean and Chinesepopulations would advance our understanding of the genetic diversity and structure ofin East Asia. In this study, we investigated the po- pulation genetics of six geographic populations, including the Bohai Sea, the Yellow Sea, the South China Sea, and the Korean Peninsula, using seven polymorphic SSR mar- kers. We also evaluated whether geographical distance af- fected the genetic structure ofat different loca- tions. This study provides a theoretical basis for the sustain-able exploitation and utilization of this valuable fishery re- source.
2 Materials and Methods
2.1 Sample Collection and Preparation
Samples were collected from six locations, including the Bohai Sea: Tianjin (TJ), Qinhuangdao (QHD); the Yellow Sea: Dandong (DD); the southwest coast of the Korean Peninsula: Mokpo (MP), Kunsan (KS); and the southwest coast of the Taiwan Strait: Penghu (PH). The detailed sam- ple information is shown in Fig.1 and Table 1. Genomic DNA was isolated frommantle muscle using an improved cetyl trimethyl ammonium bromide method (Winnepenninckx., 1993).
2.2 SSR-Enriched Sequences
The transcriptome library was constructed, and trans- criptome sequencing was performed by Gene Denovo Bio- technology Co. (Guangzhou, China) using the Illumina Hi-SeqTM 4000 (Xu and Zheng, 2020). MISA v2.1 (http://pgrc. ipk-gatersleben.de/misa/) was used to search all single gene clusters and identify the localization and type of microsa- tellites in the transcriptome following default parameters. Primers were designed using Primer3 (version 1.1.4).
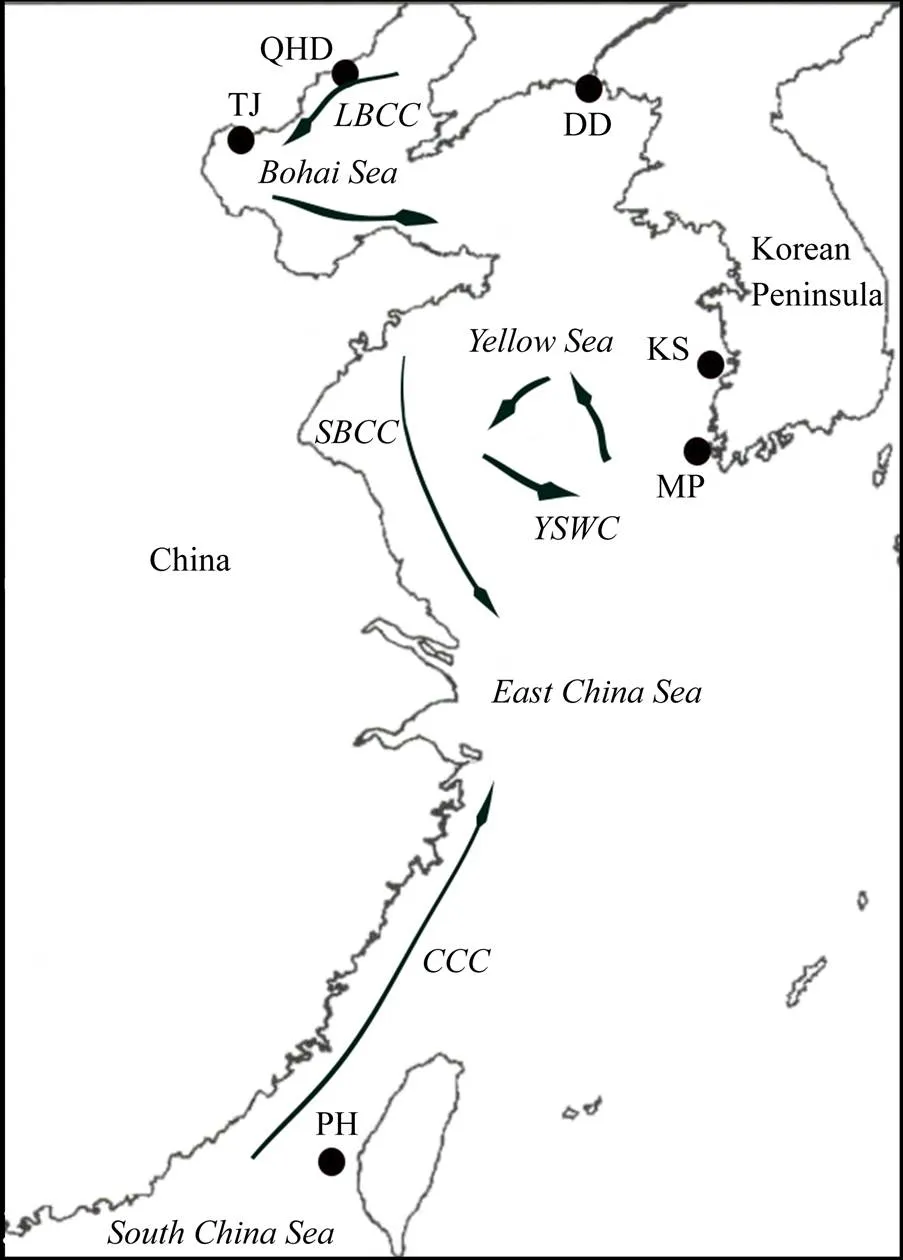
Fig.1 Map of the sampling locations and ocean currents. LBCC, Lubei Coastal Current; SBCC, Subei Coastal Cur-rent; CCC, China Coastal Current; YSWC, Yellow Sea Warm Current (Kaneko et al., 2011).
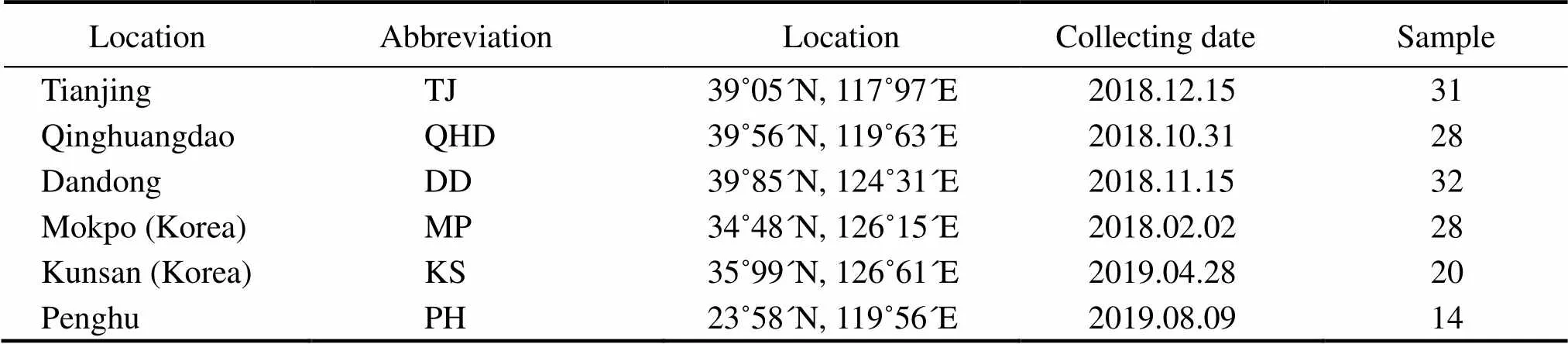
Table 1 Details of the six O. minor sampling locations
2.3 Specific Primers for SSR Loci Screening and SSR Genotyping
A total of 108 primers were selected to test polymer-phism. The polymerase chain reaction (PCR) was perform- ed in a 10μL volume, including 1μL of 1× PCR Buffer (Mg2+plus), 1μL of 0.2mmolL−1dNTP mix, 0.22μL of 1mmolL−1fluorescent label (NED, VIC, or FAM), 0.1μL of the 1mmolL−1 M13 upstream primer (F), 0.22μL of the 1mmolL−1downstream primer (R), 0.05μL of 0.25UDNA polymerase, 1μL of the 50ng DNA template, and 6.41μL of dH2O. The DNA was amplified at 94℃ for 3min, followed by 35 cycles of 94℃ for 30s, optimal an- nealing temperature for 1min, 72℃ for 75s, and then eight cycles of 94℃ for 30s, 53℃ for 1min, 72℃ for 75s, and 72℃ for 10min. Microsatellite polymorphisms were screen- ed using the ABI 3730xl DNA Analyzer.
The genotypes of the samples were analyzed at seven polymorphic microsatellite loci: OMS41, OMS44, OMS51, OMS78, OMS80, OMS96, and OMS99 (Table 1).
2.4 Statistical Analysis
Micro-Checker 2.2.3 (Oosterhout., 2004) was used to inspect the null alleles.GenALEx v.6.5 (Peakall.,2012) was used to calculate the Hardy–Weinberg equili- brium (-HWE), the number of alleles per locus (a), ob- served heterozygosity (o), expected heterozygosity (e),thestvalues,and Nei’s genetic distance. Cervus v.3.0.3 (Marshall., 1998) was used to estimate the polymor- phism information content (PIC). The neutrality of the po- lymorphic loci was analyzed using the Ewens–Watterson test in POPGENE v.1.32. Multilocus analysis of molecu- lar variance (AMOVA) and pairwisest,values were explored with ARLEQUIN v.3.5 (Excoffier and Lischer, 2010). A Mantel test implemented in Genepop (https:// genepop.curtin.edu.au/) was performed to test the isola- tion of distance (IBD) model by correlating geographic dis- tance to genetic distance (st/(1−st); Rousset, 1997). An unweighted pair group method with arithmetic mean (UP- GMA) tree was constructed based on the genetic distances among the samples from 6 locations using MEGA v.6.0.
3 Results
3.1 Characterization of the Genic Microsatellites
A total of 46192 SSR loci were discovered in 70174 uni- genes from the transcriptome, and the frequency of SSR occurrence in the unigenes was 65.82%. The best repre- sented microsatellite categories were di-nucleotide (45.26%)and mono-nucleotide (39.40%), followed by tri-nucleotideSSRs (14.94%), while tetra-nucleotide and penta-nucleotide SSRs comprised <1%. Among the dinucleotiderepeats, the most abundant repeat motif was AT/AT (22.41%), fol- lowed by AC/GT (16.49%) (Table 2). Repeating units be- tween 4 and 14 were detected in the SSRs of thetranscriptome, accounting for 99.97% of the total number, and the highest proportion of repeated times (including mo- no-nucleotide and penta-nucleotide) was six (31.89%).
3.2 Specific Primers for SSR Loci Screening
In this study, 21 of the 108 analyzed markers (19.44%) were polymorphic (showed in Table 3). The amplicon se- quences were deposited in the GenBank database (acces- sion numbers: KX061842–KX061864). The number of al- leles per loci ranged from 3 to 12, with an average of 5.6. Three loci (OMS78, OMS64, and OMS34) deviated sig- nificantly from the-HWE after a Bonferroni correction (<0.05).oranged from 0.267 to 0.941 (mean=0.614).eranged from 0.242 to 0.868 (mean=0.619). The PIC ranged from 0.231 to 0.854 (mean=0.572).The Ewens- Watterson neutral test showed that 21 microsatellite loci were located within the 95% confidence interval (Obs. F>L95), indicating the neutrality of these polymorphic markers.
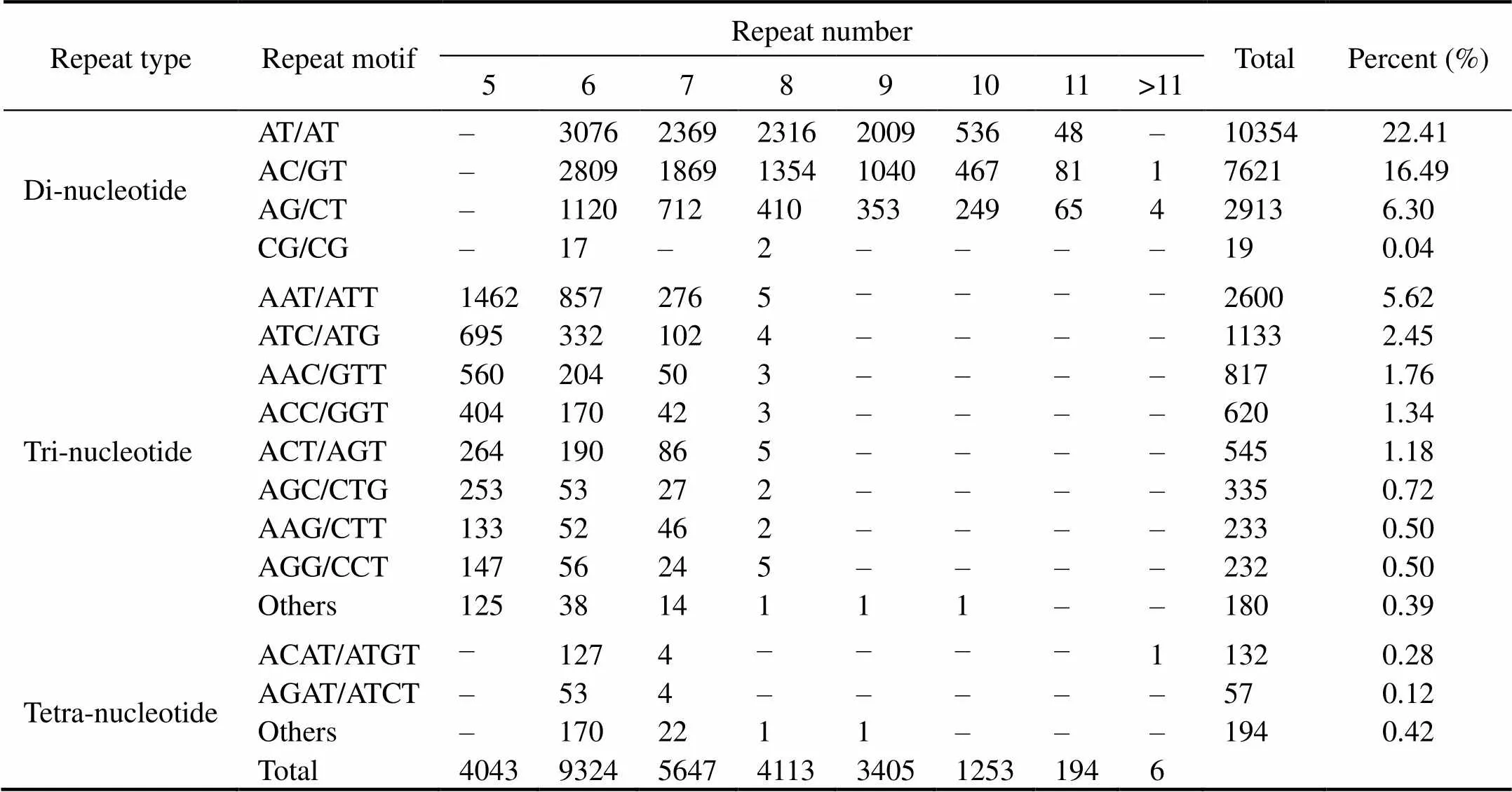
Table 2 Frequency of di-, tri-, and tetra-nucleotide repeat motifs in the transcriptome (–, not available)
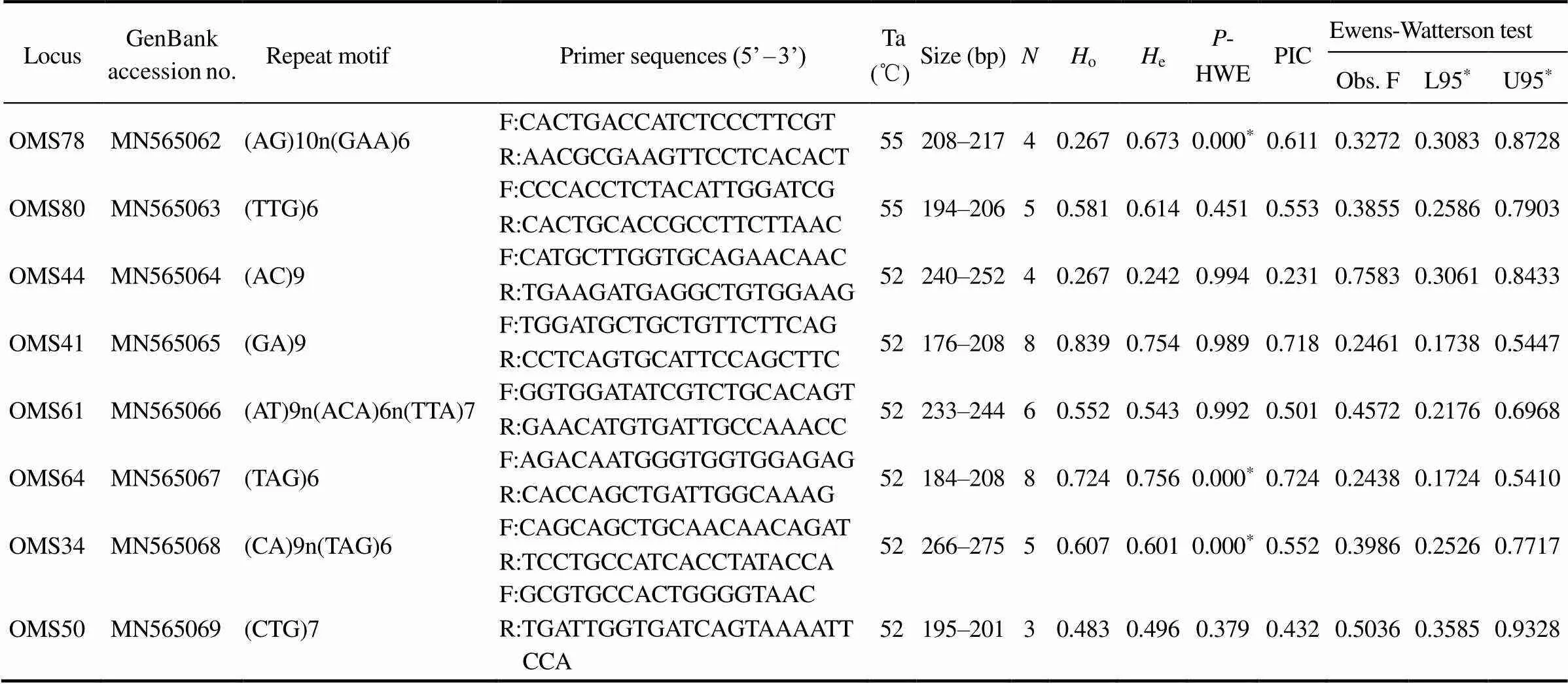
Table 3 Basic genetic information of the 21 microsatellite primers
()
()
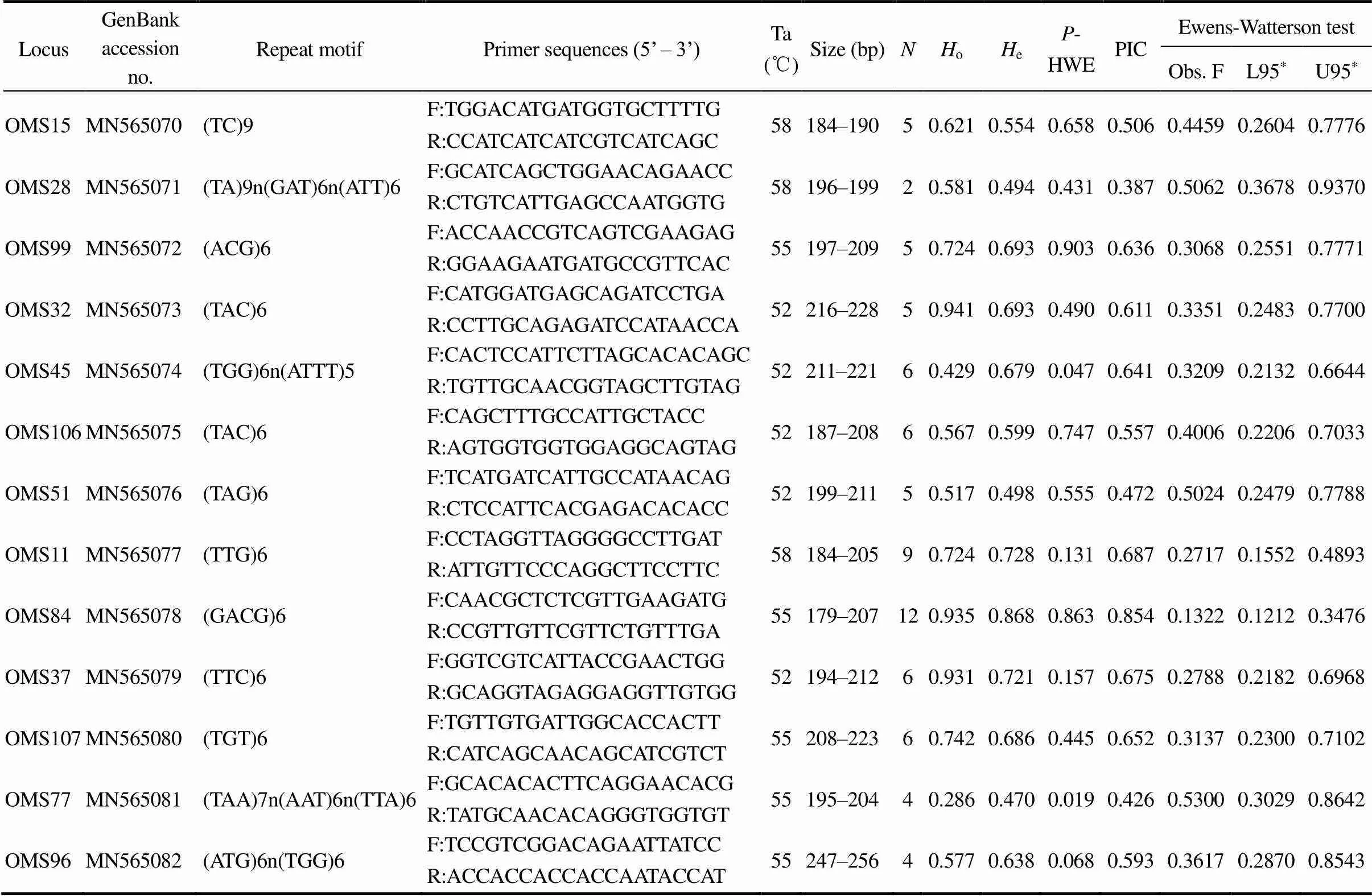
LocusGenBank accessionno.Repeat motifPrimer sequences (5’ – 3’)Ta (℃)Size (bp)NHoHeP-HWEPICEwens-Watterson test Obs. FL95*U95* OMS15MN565070(TC)9F:TGGACATGATGGTGCTTTTG58184–19050.6210.5540.6580.5060.44590.26040.7776 R:CCATCATCATCGTCATCAGC OMS28MN565071(TA)9n(GAT)6n(ATT)6F:GCATCAGCTGGAACAGAACC58196–19920.5810.4940.4310.3870.50620.36780.9370 R:CTGTCATTGAGCCAATGGTG OMS99MN565072(ACG)6F:ACCAACCGTCAGTCGAAGAG55197–20950.7240.6930.9030.6360.30680.25510.7771 R:GGAAGAATGATGCCGTTCAC OMS32MN565073(TAC)6F:CATGGATGAGCAGATCCTGA52216–22850.9410.6930.4900.6110.33510.24830.7700 R:CCTTGCAGAGATCCATAACCA OMS45MN565074(TGG)6n(ATTT)5F:CACTCCATTCTTAGCACACAGC52211–22160.4290.6790.0470.6410.32090.21320.6644 R:TGTTGCAACGGTAGCTTGTAG OMS106MN565075(TAC)6F:CAGCTTTGCCATTGCTACC52187–20860.5670.5990.7470.5570.40060.22060.7033 R:AGTGGTGGTGGAGGCAGTAG OMS51MN565076(TAG)6F:TCATGATCATTGCCATAACAG52199–21150.5170.4980.5550.4720.50240.24790.7788 R:CTCCATTCACGAGACACACC OMS11MN565077(TTG)6F:CCTAGGTTAGGGGCCTTGAT58184–20590.7240.7280.1310.6870.27170.15520.4893 R:ATTGTTCCCAGGCTTCCTTC OMS84MN565078(GACG)6F:CAACGCTCTCGTTGAAGATG55179–207120.9350.8680.8630.8540.13220.12120.3476 R:CCGTTGTTCGTTCTGTTTGA OMS37MN565079(TTC)6F:GGTCGTCATTACCGAACTGG52194–21260.9310.7210.1570.6750.27880.21820.6968 R:GCAGGTAGAGGAGGTTGTGG OMS107MN565080(TGT)6F:TGTTGTGATTGGCACCACTT55208–22360.7420.6860.4450.6520.31370.23000.7102 R:CATCAGCAACAGCATCGTCT OMS77MN565081(TAA)7n(AAT)6n(TTA)6F:GCACACACTTCAGGAACACG55195–20440.2860.4700.0190.4260.53000.30290.8642 R:TATGCAACACAGGGTGGTGT OMS96MN565082(ATG)6n(TGG)6F:TCCGTCGGACAGAATTATCC55247–25640.5770.6380.0680.5930.36170.28700.8543 R:ACCACCACCACCAATACCAT
Notes: Ta, annealing temperature;o, observed heterozygosity;e, expected heterozygosity; PIC, polymorphism information content.*Sig- nificant deviations from-HWE after Bonferroni correction (<0.05;=21).
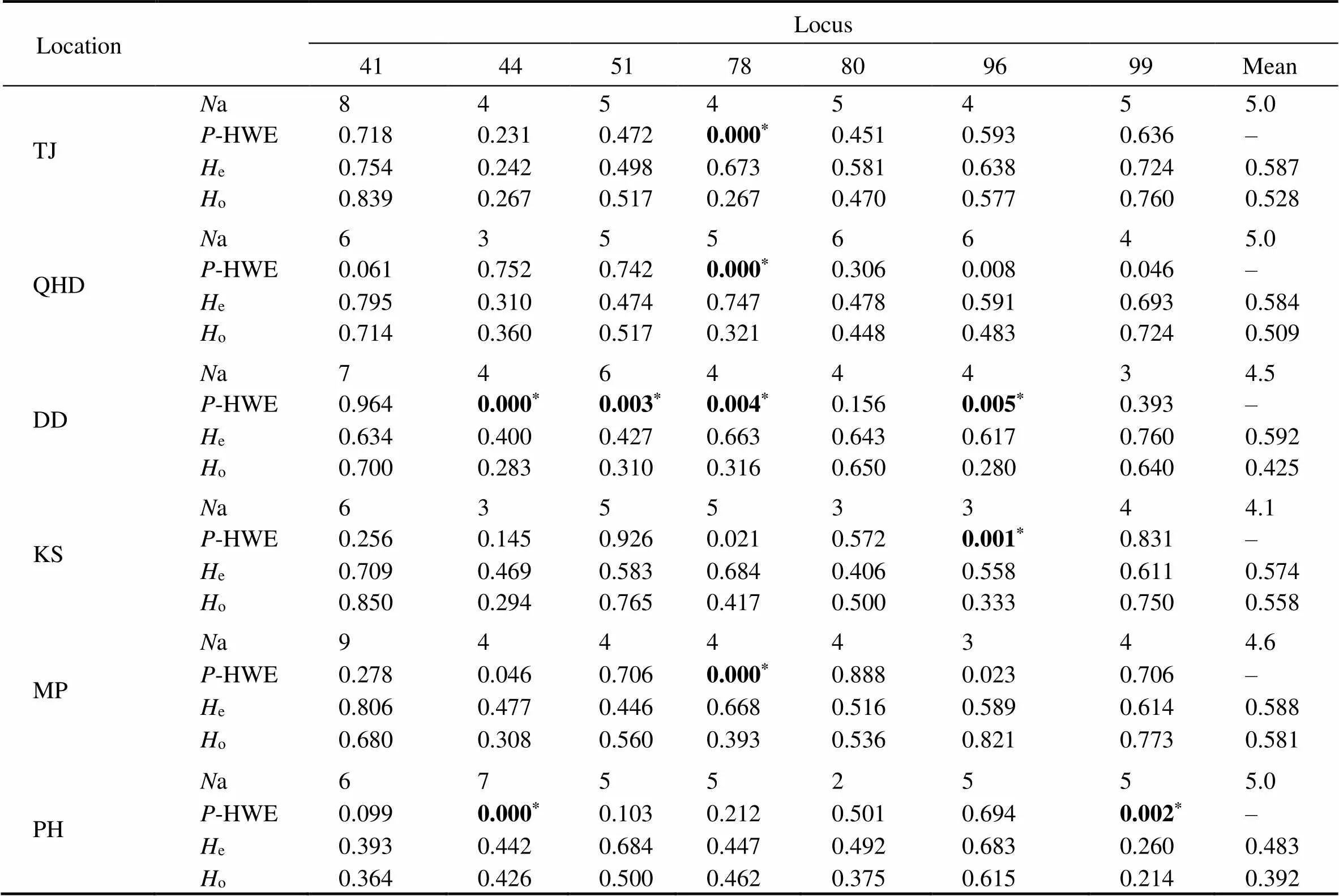
Table 4 O. minor genetic diversity indices
Notes: Ta, annealing temperature;o, observed heterozygosity;e, expected heterozygosity; R, allelic size range;*Significant deviations from the-HWE after Bonferroni correction (<0.05;=7); –, not available.
Table 4 summarizes the genetic diversity indices of 7microsatellite loci from the six sampling locations. The number of alleles per locus varied from 5 (at OMS99) to 11 (at OMS11). Observed heterozygosity values ranged from 0.241 to 0.850, and expected heterozygosity values ranged from 0.242 to 0.860. Among all loci, the average number of alleles for each location varied from 4.1 to 5.0. The lowest average number of alleles was detected in the DD (4.1), whereas TJ, QHD, and PH showed an average of five alleles per location. The average observed and ex- pected heterozygosity value per location ranged from 0.392(PH) to 0.581 (MP), and from 0.483 (PH) to 0.592 (DD), respectively. No linkage disequilibrium was detected in the locus pairs, suggesting that the loci can be treated as in-dependent variables. After the Bonferroni correction, ten of42 locus-location combinations significantly deviated from-HWE (<0.05), among which OMS78 showed a devia- tion at all four locations.
3.3 Genetic Diversity and Structure Among the Samples
Table 5 lists the Nei’s genetic distance (Dc, above thediagonal) and pairwisestvalues (below the diagonal) in pairwise comparisons at different locations. Pairwisestvalues ranged from 0.014 to 0.174, with relatively high va- lues being detected between PH and the other locations (Ta- ble 5). Similarly, the genetic distances between PH and the other locations were much larger than the pairwise com- parisons between the other locations. These results were further confirmed by a UPGMA dendrogram, which show- ed three distinct clades: a single clade for PH, one for the Korean samples (MP and KS), and one for the northern Chinese samples (TJ, QHD, and DD) (Fig.2).
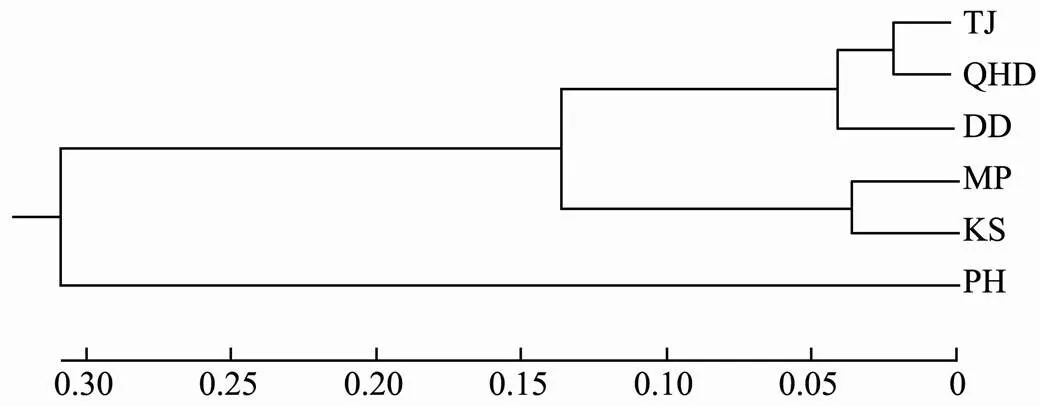
Fig.2 UPGMA tree based on matrices of the pairwise Nei’s genetic distances of the microsatellites.
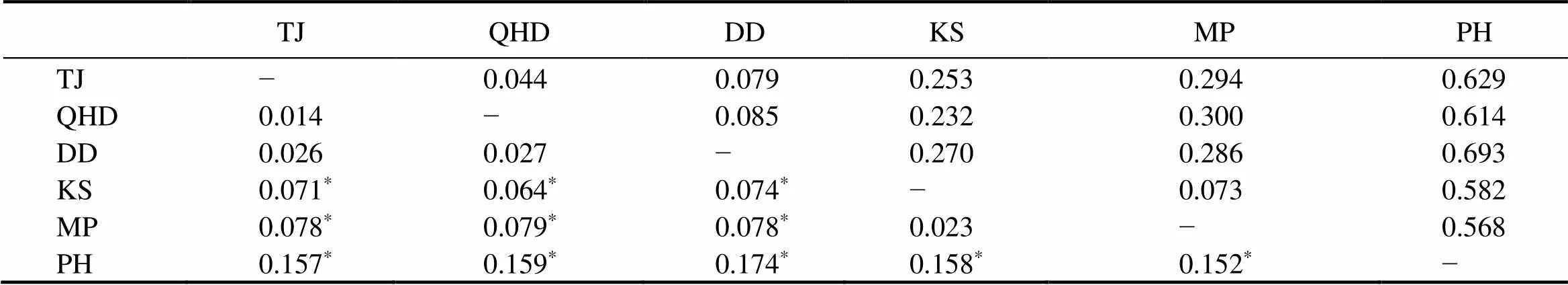
Table 5 Nei’s genetic distance (Dc, above the diagonal) and pairwise Fst values (below the diagonal) among the six O. minor locations
Note:*Significant difference after the Bonferroni correction.
3.4 Analysis of Molecular Variance and IBD
AMOVA indicated that 12% of the total genetic varia- tion occurred among the populations in the six sampling lo- cations; 26% was attributed to variation among individu- als and 63% occurred within individuals (Table 6). Accord- ing to the IBD analysis, the genetic and geographic dis- tances were highly positively correlated, and the genetic dis- tances explained 80% of the total variance (2=0.7906,=0.001), as shown in Fig.3.
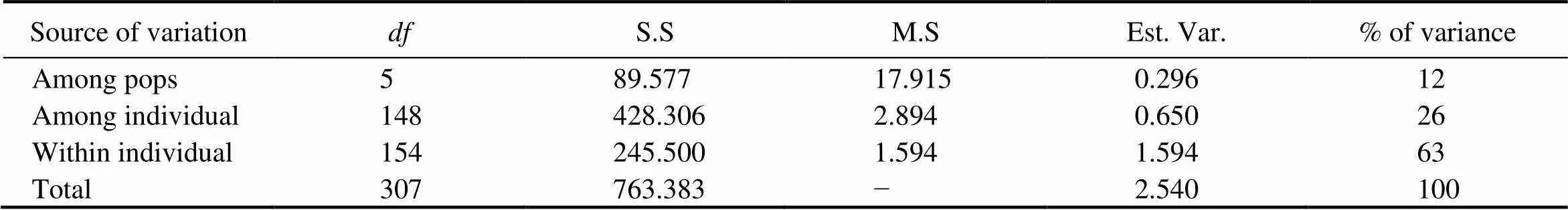
Table 6 Analysis of molecular variance (AMOVA) of genetic differentiation in O. minor (–, not available)
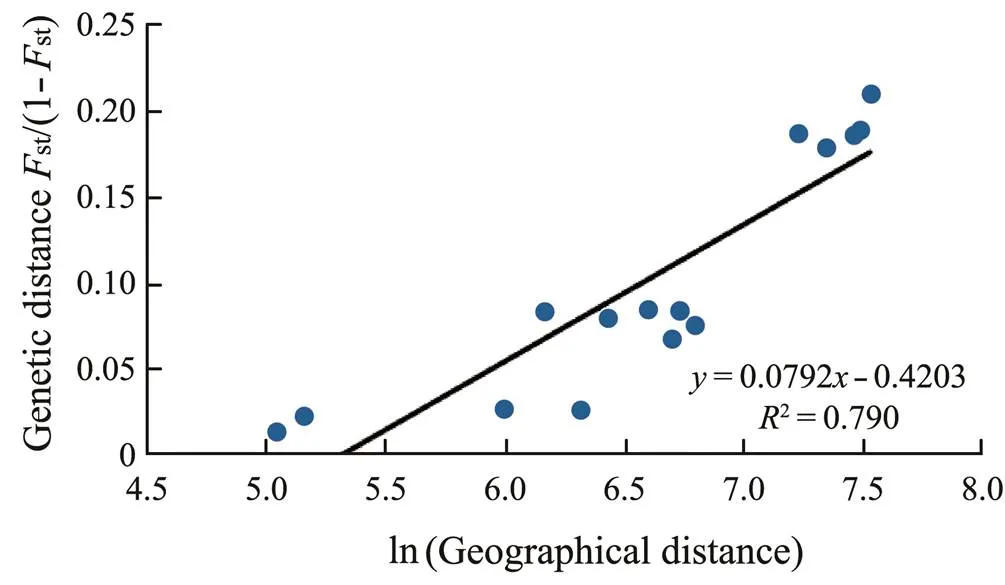
Fig.3 Scatter plot of the genetic and geographical distances for the pairwise location comparisons.
4 Discussion
A total of 46192 SSR loci inwas discovered in 70174 unigenes, and the frequency of SSR occurrence in the unigenes was 65.82%, which was much higher than that reported in(48.70%) (Guan., 2018). Mono-nucleotide and di-nucleotide SSRs accoun- ted for 39.40% and 45.26%, respectively. Similarly, di-nu- cleotides are highly over-represented in thegenome (Wang., 2018). Repeating units be- tween 4 and 14 occurred in theSSRs, account- ing for 99.97% of the total, and the highest number of re- peats was six (31.89%). The number of markers decreased as the number of repeated units and the number of repeti- tions increased, which was also observed by Sun(2017) and Shang. (2019). The markers developed in this study were highly polymorphic, as 76% of the PIC was>0.5. These highly polymorphic markers can be applied to subsequent population genetics studies.
We investigated the genetic structure offrom six geographic locations using seven microsatellite DNA markers. The average number of alleles in the different geographic populations ranged from 4.1 to 5.0, which was slightly lower than the number reported by other studies (Kang., 2012; Gao., 2016). This difference can be explained in two ways: 1) variations among the different SSR markers used in the studies; and 2) differences in the diversity and abundance of the different sampling locations. Ten of the 42 locus-location combinations significantly deviated from-HWE after a Bonferroni correction (<0.05). Kang(2012) and Gao(2016) reported that 23 of 56 and 20 of 80 locus-location combinations deviated from HWE, respectively.In addition, si- milar results have been reported in other cephalopods (Ca- branes., 2008; Doubleday., 2009). This devia-tion may be due to the small population or sample size, in-breeding, or the presence of null alleles, as indicated byother studies (Shaw., 1999; Perez-Losada., 2002; Kang., 2012).
The UPGMA tree based on Nei’s genetic distance sug- gests that samples could be arranged in three clusters. The Korean samples formed a sister clade to the samples from the Bohai Sea and the Yellow Sea, while PH samples fromthe South China Sea were separated from the other samples. PH had the largest geographical distance to the other lo- cations and the samples from PH presented the highest ge- netic distance compared to the samples from the other lo- cations. Moreover, the Korean samples (MP and KS) were also different from those of the three northern Chinese samples(TJ, QHD, and DD). Similarly, thestvalues be- tween PH and the other locations were the highest among all the comparisons. The Korean samples also had a highstvalue compared to the samples from northern China, but thestand the genetic distance values between sam-ples from the two Korean locations were relatively low. Taken together, our results indicate the divergence in the populations containing the northern Chinese samples (TJ, QHD, and DD), the Korean samples (MP and KS), and the southern Chinese sample (PH). Our results are consistent with the findings in other studies using mitochondrial and morphological markers, which reported large divergences between northern and southern populations (Xu., 2018; Gao., 2019). The population divergences between the Korean and northern Chinese samples were also revealed by Kang. (2012).
The IBD analysis in Fig.3 shows a strong correlationbetween genetic distance and geographical distance, ac- counting for about 80% of the total divergence. Therefore, our results indicate that these divergence results can be mainly explained by the theory of genetic and geographi- cal distances, which hypothesizes that the effect of gene flow can lead to genetic similarities among locations with small geographical distances; therefore, geographical pro- ximity may reflect the high correlation between genetic distance and geographical distance (Scribner., 1986). However, other effects, such as ecological habitat and oceancurrents, can also contribute to population divergence. For example, Kang. (2012) suggested that divergence of theKorean population may be due to ecological differences in the habitats between the western muddy coast and the southern rocky areas. Many studies have shown that ocean currents play a crucial role in the popu- lation genetic differentiation of benthic marine organisms (Doubleday., 2009; Zhan., 2009; Ni., 2011; Gao., 2016). The Yellow Sea Warm Current flows through the west coast of the Korean Peninsula into the northwestern coast of China during April–August. How- ever, this current is relatively weak compared with the coastal waters of China and rarely reaches the inside of the Gulf of Bohai Sea (Pang and Kim, 1998). The dura- tion of the benthic planktoniclarval stage ofis relatively short (Zheng., 2014). Weinferred that geo-graphic distance is one of the main factors affecting the dispersal ofAlthough other factors might simul- taneously affect genetic differentiation, the IBD model ex- plained about 80% of the variance of the six geographical populations in this study.
5 Conclusions
In the present study, 16 of 21 genetic SSR markers were polymorphic, demonstrating the feasibility and effective- ness of developing neutral and polymorphic SSRs derived fromtranscriptomic data. The genetic differentia- tion amongsampling locations based on SSRs showed that the samples from the Taiwan Strait differed greatly from the northern Chinese and Korean samples, and genetic divergence was also detected between the Chinese and Korean samples. The present results reveal valuable information to describe the genetic structure and to mo- nitordemographic parameters.These markers will be helpful to manage and conserve thefish- ery.
Acknowledgements
We thank Associate Professor Mongfang Li (National Penghu University of Science and Technology) and Dr. Hae-Li Lee (Kunsan Seafood Company) for providing the specimens. This study was supported by the National Na- tural Science Foundation of China (No31672257), and theNational Key Research and Development Program of Chi- na (No. 2020YFD0900705).
Allcock, A. L., Lindgren, A., and Strugnell, J. M., 2015. The con- tribution of molecular data to our understanding of cephalo- pod evolution and systematics: A review., 49 (21-24): 1373-1421.
Barco, A., Raupach, M. J., Laakmann, S., Neumann, H., and Knebelsberger, T., 2016. Identification of North Sea mollusks with DNA barcoding., 16 (1): 288-297.
Cabranes, C., Fernandez-Rueda, P., and Martinez, J. L., 2008. Genetic structure ofaround the Iberian Pe- ninsula and Canary Islands as indicated by microsatellite DNA variation., 65: 12-16.
Chen, Z. W., Xu, R., Nan, Z., and Zheng, X. D., 2019. Determi- nation of the median lethal concentration and acute toxicity ofunder different concentrations of ammonia andnitrogen stress., 50 (6): 1361-1370 (in Chinese with English abstract).
Doubleday, Z. A., Semmens, J. M., Smolenski, A. J., and Shaw, P. W., 2009. Microsatellite DNA markers and morphometrics reveal a complex population structure in a merobenthicspecies () in south-east Australia and New Zealand., 156 (6): 1183-1192.
Excoffier, L., and Lischer, H. E. L., 2010. Arlequin suite ver 3.5: A new series of programs to perform population genetics ana- lyses under Linux and Windows., 10: 564-567.
Gao, X., Xu, R., Zhang, Z., and Zheng, X., 2019. Morphological variation analysis ofin the coastal waters ofChina., 43 (7): 1593-1602 (in Chinese with English abstract).
Gao, X., Zheng, X., Bo, Q., and Qi, L., 2016. Population geneticsof the common long-armed octopus(Sasaki,1920) (Cephalopoda: Octopoda) in Chinese waters based onmicrosatellite analysis.,66: 129-136.
Guan, A., Wu, Y., Chen, Y., Sun, Y., Qi, P., and Guo, B., 2018. Deep sequence-based transcriptome analysis of microsatelli- tes in the cuttlefish ()., 39: 144-151 (in Chinese with English abstract).
Kaneko, N., Kubodera, T., and Iguchis, A., 2011. Taxonomic study of shallow-water octopuses (Cephalopoda: Octopodidae) in Ja- pan and adjacent waters using mitochondrial genes with per- spectives on octopus DNA barcoding., 54 (1-2): 97-108.
Kang, J. H., Kim, Y. K., Park, J. Y., An, C. M., and Jun, J. C., 2012. Development of microsatellite markers to genetically diffe- rentiate populations offrom Korea and China., 39: 8277-8286.
Kim, D. H., An, H. C., Lee, K. H., and Hwang, J. W., 2008. Op- timal economic fishing efforts in Korean common octopustrap fishery., 74: 1215-1221.
Marshall, T. C., Slate, J., Kruk, L. E., and Pemberton, J. M., 1998. Statistical confidence for likelihood-based paternity in- ference in natural populations., 7: 639-655.
Moreira, A. A., Tomas, A. R. G., and Hilsdorf, A. W. S., 2011. Evi- dence for genetic differentiation of(Mol-lusca, Cephalopoda) fishery populations from the southern coastof Brazil as revealed by microsatellites., 407 (1): 34-40.
Ni, L. H., Li, Q., and Kong, L. F., 2011. Microsatellites reveal fine-scale genetic structure of the Chinese surf clam(Mollusca, Bivalvia, Mactridae) in Northern China., 32 (4): 488-497.
Oosterhout, C. V., Hutchinson, W. F., Wills, D. P. M., and Ship- ley, P., 2004.MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data., 4 (3): 535-538.
Pang, I. C., and Kim, K. H., 1998. Seasonal variation of water mass distributions in the eastern Yellow Sea and the Yellow Sea Warm Current., 3 (3): 41-52.
Peakall, R., and Smouse, P. E., 2012. Genalex 6.5: Genetic ana- lysis in Excel. Population genetic software for teaching and research–An update., 28: 2537-2539.
Perez-Losada, M., Guerra, A., Carvalho, G. R., Sanjuan, A., and Shaw, P. W., 2002. Extensive population subdivision of the cuttlefish(Mollusca: Cephalopoda) around the Iberian Peninsula indicated by microsatellite DNA variation., 89: 417-424.
Rousset, F., 1997. Genetic differentiation and estimation of gene flow from-statistics under isolation by distance.,145: 1219-1228.
Scribner, K. T., Evans, J. E., Morreale, S. J., and Smith, M. H., and Gibbons, J. W., 1986. Genetic divergence among popula- tions of the yellow-bellied slider turtle () se- parated by aquatic and terrestrial habitats., 3: 691-700.
Seol, D. W., Lee, J., and Im, S. Y., 2007. Clove oil as an anes- thetic for common octopus (, Sasaki)., 38 (1): 45-49.
Shang, G. J., Xu, A., Hu, X., and Li, Z., 2019. Isolation and cha- racterization of genic microsatellites fromassembly transcriptome in the bivalve, 37: 1071-1079.
Shaw, P., Pierce, J., and Boyle, P. R., 1999. Subtle population structuring within a highly vagile marine invertebrate, the vein-ed squid, demonstrated with microsatellite DNA markers., 8: 407-417.
Song, M. P., Wang, J. H., and Zheng, X. D., 2019. Prey prefer- ence of the common long-armed octopus(Ce- phalopoda: Octopodidae) on three different species of bivalves., 37 (5): 1595-1603.
Sun, B. C., Yang, J. M., Sun, G. H., Liu, X. Q., Liu, L. J., Wang, W. J.,., 2010. Sequence and molecular phylogeny of mitochondrial COI gene fragment in five populations ofin China., 41 (2): 259-265 (in Chinese with English abstract).
Sun, X., Li, D., Liu, Z., Zhou, L., Wu, B., and Yang, A., 2017.assembly of pen shell () transcrip- tome and screening of its genic microsatellites., 16: 882-888.
Tang, Y., Zheng, X., Liu, H., and Sun, F. G., 2020. Population ge- netics and comparative mitogenomic analyses reveal cryptic diversity of(Cephalopoda: Octopodi- dae)., 112 (6): 3893-3902.
Wang, J. H., and Zheng, X. D., 2017. Comparison of the genetic relationship between nine cephalopod species based on clus- ter analysis of karyotype evolutionary distance., 11 (3): 477-494.
Wang, L., Yu, H., and Li, Q., 2018. Identification and characterization of 23 microsatellite loci forbased on RAD-seq.,97 (1): 61-65.
Winnepenninckx, B., Backeljau, T., and Wachter, R. D., 1993. Extraction of high molecular weight DNA from molluscs., 9 (12): 407.
Xu, R., and Zheng, X. D., 2020. Hemocytes transcriptomes re- veal metabolism changes and detoxification mechanisms in response to ammonia stress in., 29 (9): 1441-1452.
Xu, R., Bo, Q. K., and Zheng, X. D., 2018. A divergent lineage among(Sasaki, 1920) populations in the North- west Pacific supported by DNA barcoding., 14 (4): 335-349.
Yamamoto, T., 1942. On the ecology of(Sasaki), with special reference to its breeding habits., 12: 9-20.
Zhan, A. B., Hu, J. J., Hu, X. L., Zhou, Z. C., Hui, M., Wang, S.,., 2009. Fine-scale population genetic structure of Zhikong scallop (): Do local marine currents drive geo- graphical differentiation?, 11 (2): 223- 235.
Zheng, X. D., Qian, Y. S., Liu, C., and Li, Q., 2014.. In:Iglesias, P.,., eds., Springer, New York, 415-426.
Zuo, Z. R., Zheng, X. D., Yang, Y., and Li, Q., 2011. Development and characterization of 12 polymorphic microsatellite loci in(Sasaki, 1920).,3: 489-491.
February 1, 2021;
May 24, 2021;
November 11, 2021
© Ocean University of China, Science Press and Springer-Verlag GmbH Germany 2022
. Tel: 0086-532-82032873
E-mail:xdzheng@ouc.edu.cn
(Edited by Qiu Yantao)
杂志排行

Journal of Ocean University of China的其它文章
- Variations in Dissolved Oxygen Induced by a Tropical Storm Within an Anticyclone in the Northern South China Sea
- Intensity of Level Ice Simulated with the CICE Model for Oil-Gas Exploitation in the Southern Kara Sea, Arctic
- Learning the Spatiotemporal Evolution Law of Wave Field Based on Convolutional Neural Network
- Development and Control Strategy of Subsea All-Electric Actuators
- Acoustic Prediction and Risk Evaluation of Shallow Gas in Deep-Water Areas
- In Situ Observation of Silt Seabed Pore Pressure Response to Waves in the Subaqueous Yellow River Delta