短期牦牛放牧对青藏高原高寒草地土壤真菌群落的影响
2022-10-24王永宏田黎明艾鷖陈仕勇泽让东科
王永宏,田黎明,艾鷖,陈仕勇,泽让东科*
(1.西南民族大学青藏高原研究院,四川若尔盖高寒湿地生态系统国家野外科学观察研究站,四川成都 610041;2.四川大学生命科学学院,四川成都 610065;3.西南民族大学畜牧兽医学院,四川 成都 610041)
青藏高原是全球海拔最高、面积最大的高原,具有极其重要的生态价值[1]。高寒草地约占青藏高原总面积的60%[2],是全球最独特的草地生态系统之一,在保护青藏高原生物多样性方面发挥着重要的生态作用,同时维持着区域的经济发展[3]。青藏高原是我国主要牧区之一[4],放牧作为主要的草地利用方式,对青藏高原的经济发展与生态环境具有重要影响[5]。然而,随着放牧强度严重超载,加之气候变暖,最终导致青藏高原高寒草甸发生了不同程度的退化[6-7]。持续高强度放牧会对草地造成不可恢复的损害,但单一限制牧民放牧会阻碍青藏高原的经济发展[8]。因此,探究青藏高原地区牦牛放牧、经济发展与草地生态健康的平衡具有重要社会意义。
微生物是生态系统中主要的分解者之一,微生物多样性与生态系统功能直接相关,微生物丰富度越高,群落功能越稳定[9]。放牧通过采食、践踏与排泄行为影响土壤性质和微生物群落,家畜会通过排泄方式返还给土壤部分碳[10],家畜的消化与分解过程,会使排泄物结构变简单,从而利于微生物的利用与生长。放牧行为会影响土壤真菌群落的结构,放牧强度也会对微生物群落产生极大的影响。放牧强度过高会改变土壤的容重,降低植被覆盖度,增加土壤表面的紫外线强度,改变土壤温度,从而使微生物的生长条件恶化[11],导致微生物生物量下降。土壤真菌的底物为难分解的有机质,在放牧地的土壤微生物群落竞争中处于劣势,土壤微生物群落向以细菌为主的结构转变[12]。但Wang等[13]通过添加不同形式的碳发现真菌主要同化不稳定碳,虽然对葡萄糖利用能力不及细菌,但当葡萄糖碳可用性增加时,真菌是固定葡萄糖碳的关键竞争类群。放牧通过多种方式影响土壤真菌群落,但牦牛放牧强度对于真菌群落的影响仍不清楚。
全球范围的研究表明,土壤真菌的丰富度和植物多样性仅存在微弱的关系,并提出气候因素、土壤和空间变量是真菌丰富度与群落结构的最佳预测因子[14]。针对青藏高原的研究表明在考虑环境因子时真菌丰富度与植物丰富度呈正相关,但气候、空间和土壤仍是真菌群落组成的主要预测因子[15]。放牧作为草地利用的主要形式之一,许多研究揭示了其对真菌群落的影响[16-19]。例如,青藏高原围封试验发现放牧活动会降低土壤有机碳,间接增加真核生物的丰富度与多样性[16];究其原因主要是放牧通过减少土壤碳输入使土壤pH升高,从而为真菌生长提供更适合的条件,使土壤真菌的α多样性与土壤pH呈正相关[17]。将真菌根据功能群进一步划分后发现,不同的真菌功能群对放牧的响应并不相同:放牧会增加真菌粪腐菌、降低植物病原菌,兔子放牧降低了外生菌根和丛枝菌根真菌[18-19];通过全球范围草地的整合分析发现重度和中度放牧会降低丛枝菌根真菌丰度,而轻度放牧的影响却不显著[20]。前期研究表明土壤因子是放牧影响土壤真菌的重要媒介之一,但放牧对土壤的影响具有一定的滞后性,因此短期放牧对土壤化学计量的影响并不显著[21],然而短期放牧对土壤真菌是否有影响及其影响方式报道较少。
真菌群落的生态功能,是通过物种之间的相互作用实现的,物种间的竞争、协作等过程直接或间接相互联系,形成一个复杂的互作网络[22]。网络分析能够通过模块化量化共现网络的相互作用程度,进一步细分了真菌类群的生态功能[23]。以往的研究中将真菌群落作为一个整体,探究其多样性及丰度对放牧活动的响应;或者仅研究特定的功能群对于放牧的响应,忽略了不同真菌间的竞争与合作关系。真菌群落拥有复杂的结构,不同真菌类群间具有怎样的相互作用及其相互作用是否会削弱特定真菌类群对外界干扰的响应仍需进一步探究。
本研究在川西北高寒草甸进行短期(2年)不同强度牦牛放牧处理,利用二代测序技术对土壤真菌群落结构进行探究,通过不同真菌物种的共现特征进行真菌类群划分,研究牦牛短期放牧对土壤真菌群落构成是否有影响,若有影响是通过何种方式实现的,从而为高寒草甸放牧管理提供基础数据与依据。
1 材料与方法
1.1 试验地概况
本试验在“四川若尔盖高寒湿地生态系统国家野外科学观察研究站”进行,位于青藏高原东缘四川省阿坝藏族羌族自治州(32°48'N,102°33'E),试验区年均气温1.5℃、年均降水750 mm,海拔为3504 m。试验地优势植物种为垂穗披碱草(Elymus nutans)、高山嵩草(Kobresia pygmaea)、矮嵩草(Kobresia humilis),植被类型为典型高寒草甸。
1.2 试验设计与样品采集
于2014年选取一处典型高寒草甸夏季牧场进行围封,围封一年确保草地初始状况较为均一,2015年5月进行放牧试验,牦牛的放牧时间为每年的5月下旬到9月下旬。试验设计为3个放牧处理和一个对照处理,每个处理设置3个重复;放牧处理为轻度放牧(1头牦牛·hm-2)、中度放牧(2头牦牛·hm-2)与重度放牧(3头牦牛·hm-2),对照处理为围封禁牧。试验地总面积为10 hm2,其中每个放牧处理样地面积为1 hm2,每个对照处理样地面积均为0.33 hm2,样地的详细情况见Mipam等[21,24]的报道。试验期间每个月对每块样地随机选取4个点,进行植物群落调查与生物量样品收集。于放牧处理第2年8月底采集土壤样品,对每块样地随机选取4个点,每个点用土钻采集5个0~10 cm的土柱混合为一个土壤样品;土壤样品分为两部分,一部分阴干用于土壤理化性质检测,另一部分低温保存用于土壤真菌群落测定。
1.3 环境因子测定
植物群落丰富度通过计算每个调查的样方的植物物种数来确定,Shannon-Wiener多样性指数和Pielou均匀度指数根据以下公式计算:
式中:H为Shannon-Wiener多样性指数,J为Pielou均匀度指数,S为物种丰富度,Pi为每个样方中的物种i的相对重要性,其计算方法为物种i的个体数与样方总个体数的比值。植物净生产力使用可移动扣笼法估计,其方法为在每个放牧区放置4个可移动扣笼,在试验期每月收获可移动扣笼内与外的生物量,并在收获完当月生物量后随机移动扣笼,通过笼内与笼外收获生物量的差值获得当月的家畜消耗量,估算各试验地净初级生产力。
采用烘干法测定土壤含水率,环刀法测定土壤容重;将阴干土壤拣除草根、石块等杂物后用研砵碾碎,过2 mm筛后用于土壤全效养分检测,采用电位法测定土壤pH,重铬酸钾氧化外加热法测定土壤有机质,凯氏定氮法测定土壤全氮,NaOH熔融-钼锑抗比色法测定土壤全磷,火焰分光光度计法测定土壤全钾;用过0.15 mm筛的阴干土壤测速效养分,采用碱解扩散法测定土壤碱解氮含量,NaHCO3浸提-钼锑抗比色法测定土壤速效磷,乙酸铵浸提-火焰光度计法测定土壤速效钾,以上所有测定方法均参照鲍士旦[25]。
1.4 土壤真菌群落测定
土壤总DNA用QIAamp® DNA Stool Mini Kit(QIAGEN,德国)试剂盒进行提取,用引物ITS5-1737F(5'-GGAAGTAAAAGTCGTAACAAGG-3')和ITS2-2043R(5'-GCTGCGTTCTTCATCGATGC-3')对真菌ITS1区域进行PCR扩增,PCR过程为:通过98℃保持30 s进行预变性,然后进入循环;循环过程为98℃变性15 s、降到50℃退火30 s、72℃延伸30 s完成循环,循环次数为25~27次;最后72℃保持5 min,于4℃保存。回收PCR产物后在Microplate reader(BioTek,FLx800)上进行定量,用Illumina公司的TruSeq Nano DNALT Library Prep Kit制备测序文库,采用Illumina MiSeq测序平台进行双端测序。
1.5 生信分析
将原始双端测序数据利用滑动窗口法进行质控,窗口设置为10 bp,步长设置为1 bp,从5'端进行窗口滑动,窗口的碱基平均测序准确率≥99%(Q20),从第一个平均质量低于该标准的窗口进行截断,去除截断后小于150 bp和含有模糊碱基N的序列。使用软件FLASH(v1.2.7)将Read1与Read2进行碱基配对,重叠碱基长度≥10 bp,最后根据Barcode序列对样本进行拆分。使用QIIME(quantitative insights into microbial ecology,v1.8.0)的USEARCH(v5.2.236)去除嵌合体序列,调用UCLUST工具,进行序列比对,将序列按照97%的相似度进行可操作分类单元(operational taxonomic unit,OTU)的划分,选取丰度高的序列作为代表序列,将代表序列与UNITE数据库比对进行分类学注释。
1.6 统计分析
利用SPSS对土壤理化性质进行单因素方差分析(P<0.05)。使用QIIME计算真菌的α多样性指数,用R的aov函数对α多样性指数进行单因素方差分析;用R的mixOmics包进行有监督判别分析,对不同放牧强度处理的组间差异进行分析,并进行可视化;使用R的Hmisc包进行所有处理OTU的相关分析,保留大于0.6的相关关系,用Gephi可视化真菌共生网络并计算模块性,并将真菌模块的相对丰度与环境因子进行相关性分析;用R的pacman包进行环境因子的筛选,将真菌门分类水平下的相对丰度用筛选出的环境因子进行冗余分析(redundancy analysis,RDA),并将预测因子进行分类,对真菌群落结构变化进行方差分解(variance partitioning analysis,VPA)。用R的ggplot 2包可视化真菌的群落组成与真菌模块结构;用R的aov函数对各模块的相对丰度进行单因素方差分析,用TukeyHSD函数进行多重比较,并可视化。
2 结果与分析
2.1 不同放牧强度对环境因子的影响
由表1可知,短期放牧处理下放牧强度间植物群落特征差异比土壤性质更大,植物多样性随放牧强度的增加而增加,中度与重度放牧显著增加了植物群落多样性(P<0.05);均匀度呈现相似的变化规律,但仅有重度放牧相比于对照显著增加了植物均匀度;放牧会增加植物群落的地上净初级生产力,并且轻度与中度放牧强度显著增加了植物群落的生产力,中度放牧的草地生产力最高。不同放牧强度的土壤容重存在显著差异,轻度放牧与重度放牧显著降低了土壤容重;土壤化学性质方面,放牧强度增加使土壤速效磷含量升高,并且重度放牧与对照相比显著增加;土壤pH在不同放牧强度间具有显著差异,放牧活动使土壤酸性增强,中度放牧的土壤pH值最小,显著小于对照组和轻度放牧。
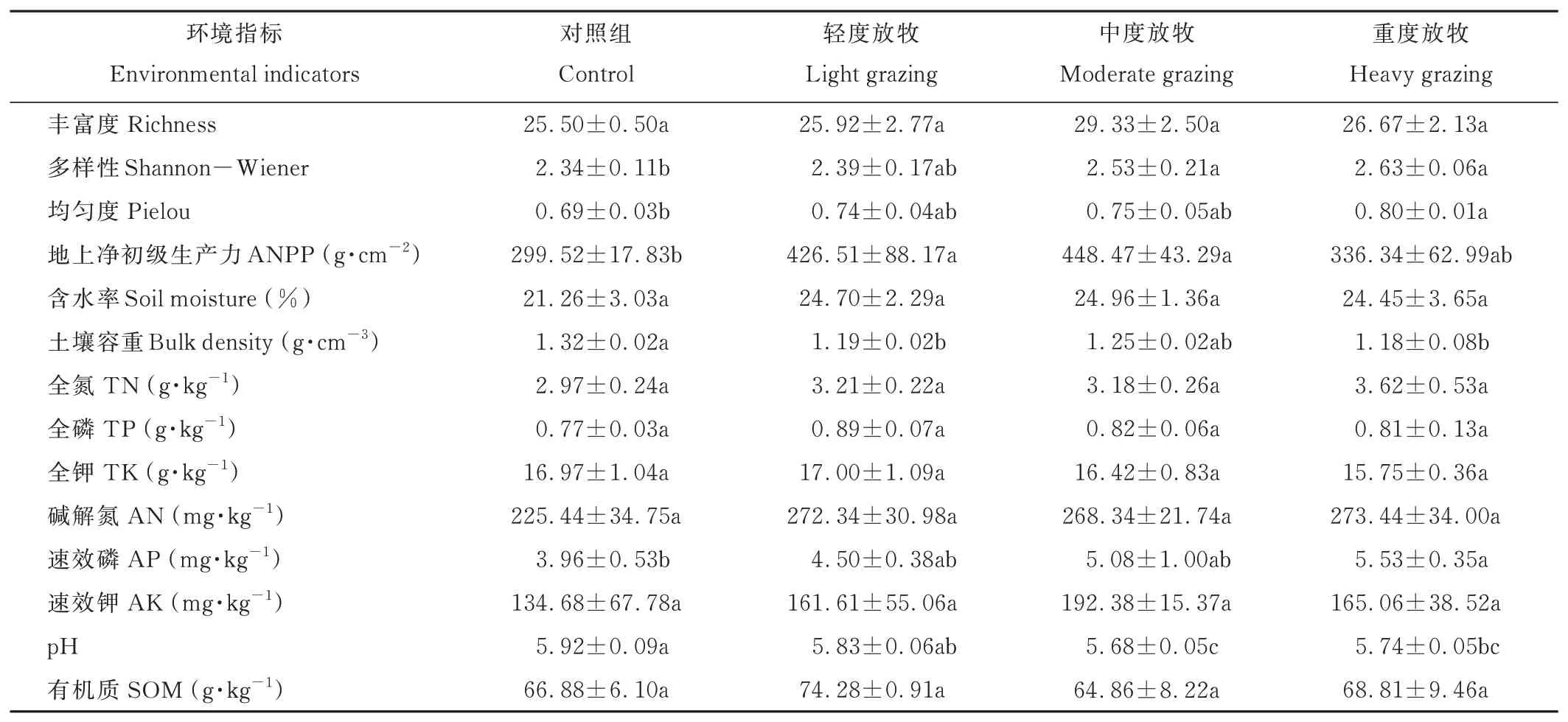
表1 不同放牧强度对环境因子的影响Table 1 Effect of different grazing intensities on environmental factors
2.2 不同放牧强度对土壤真菌群落的影响
2.2.1土壤真菌群落的组成 结果表明,土壤真菌群落主要由子囊菌门(Ascomycota)、担子菌门(Basidiomycota)和 接 合 菌 门(Zygomycota)组 成,还 包 括 少 量 的 球 囊 菌 门(Glomeromycota)、隐 真 菌 门(Rozellomycota)和壶菌门(Chytridiomycota);其中土壤中子囊菌门数量最多,轻度、中度、重度的相对丰度比对照分别增加了31.05%、7.69%、40.53%;而作为土壤真菌群落另一主要组成成分,担子菌门在轻度、中度、重度的相对丰度比对照组分别降低了37.34%、17.10%、47.42%。测序结果还包含了原生生物纤毛虫门(Ciliophora)和丝足虫门(Cercozoa)与植物界的被子门(Anthophyta),但其相对丰度值较低,不影响土壤真菌的整体群落结构(图1)。
2.2.2土壤真菌群落的α多样性 土壤真菌群落的α多样性指数在不同放牧强度中均无显著差异(表2)。Chao1丰富度指数利用群落中检测到1和2次的OTU数估算群落的物种数。Chao1丰富度指数随放牧强度呈现先增加后减少的趋势,轻度放牧的丰富度最高,中度放牧与重度放牧的土壤真菌Chao1丰富度指数值大致相同。放牧处理同样具有土壤真菌多样性增加的趋势,但重度放牧的Shannon多样性指数最大,其多样性最高。

表2 不同放牧强度土壤真菌群落α多样性的单因素方差分析结果Table 2 Results of One-way ANOVA analysis of soil fungal community α diversity at different grazing intensities
2.2.3土壤真菌群落的组间差异分析 结果表明,偏最小二乘法判别分析(partial least squares discrimination analysis,PLS-DA)的两个坐标轴的解释度分别为11.5%和11.6%;其中对照组和重度放牧两个处理的组内各样本距离近,组内重复效果好;中度放牧与对照组距离最近,其对土壤真菌群落结构的影响最小,重度放牧对土壤真菌群落的影响最为明显。对照组处于排序的中间位置,可见不同的放牧强度对土壤真菌群落结构的影响是不同的(图2)。
2.3 土壤真菌群落共现网络
基于真菌群落OTU数据相关性构建了共现网络图,共得到了308个节点、3078条边的真菌网络结构。对真菌网络进行模块划分后,得到模块化值为0.936的模块网络,并划分为4个主要的模块。模块1包含OTUs种类最多,共有94个节点,占网络总数的30.52%;其次为模块2,共有84个节点,占网络总节点数的27.27%;模块3由57个节点组成,占网络总节点数的18.51%;模块4包含了27个节点,占网络总数的8.77%。其中模块2节点连通度高,和其他节点的相互作用更强,对真菌群落的影响更加强烈(图3A)。
子囊菌门作为土壤中主要的真菌类群,也是4个模块的主要组成部分。作为组成种类最丰富的模块,模块1的子囊菌门真菌由52种OTUs组成,其在模块1的相对丰度为56.09%;而担子菌门的相对丰度为41.37%,包括31个OTUs种类,是各个模块中担子菌门相对丰度最高的模块。模块2未能识别的真菌丰度增加;但其主要组成类群子囊菌门的相对丰度为59.51%,由46种真菌组成;担子菌门包含了16种真菌,在模块2的相对丰度为12.56%。模块3则基本由子囊菌门组成,其子囊菌门包括了25种OTUs,相对丰度则为82.51%。模块4是4个主要模块结构最简单的,其子囊菌门的相对丰度也是4个模块中最少的(33.13%),仅包含11种OTUs;担子菌门的相对丰度为17.45%,由9个真菌种组成;模块4的接合菌门相对丰度相对于其他模块明显增加,虽然只包含2种OTUs,但其相对丰度为18.54%(图3B)。
2.4 不同放牧强度对土壤真菌模块的影响
模块1、模块2与模块3在不同放牧强度的相对丰度差异不显著。模块1的相对丰度随放牧强度呈现先增加后减少的趋势,模块1的轻度放牧相对丰度最高;模块3的放牧处理相对丰度均高于对照处理,并随着放牧强度的增加呈现逐渐增大的趋势;虽然模块4的相对丰度最少,但其对于放牧强度的响应最强烈,模块4的重度放牧相对丰度显著高于(P<0.1)对照组,而轻度放牧和中度放牧与对照差异不显著,与重度放牧差异也不显著(图4)。
2.5 土壤真菌与环境因子的关系
2.5.1环境因子对真菌群落的影响 通过对环境因子进行共线性排除与模型筛选,选择植物群落Shannon-Wiener多样性指数和土壤化学性质全磷、全钾、速效磷以及速效钾作为真菌门分类水平的预测因子进行冗余分析,第一、二解释轴的解释度分别为43.67%和25.02%,组成真菌群落的主要类群子囊菌门受土壤速效磷与植物群落多样性的影响,担子菌门则与土壤全钾和速效钾呈正相关关系(图5A)。进一步对真菌群落结构进行方差分解后发现,土壤因子对真菌类群变化的解释度更高,其排除植物因子的解释度为39.1%,而植物因子排除土壤的解释度为4.9%(图5B)。
2.5.2环境因子与真菌模块间的关系 由图6可知,植物群落特征与各真菌模块相关性较弱,作为真菌群落的主要组成类群,模块1与模块2对环境因子的响应是相反的,均受到土壤全磷与全钾的显著影响,土壤的全磷与全钾含量与模块1的相对丰度呈显著正相关,与模块2显著负相关。模块3是受土壤性质影响最多的真菌类群,与碱解氮、速效磷等土壤速效养分呈显著正相关,也受到土壤物理性质的影响,与土壤含水率极显著正相关,与土壤容重极显著负相关。模块4受环境影响相对较小,仅与土壤全钾呈显著负相关。
3 讨论
3.1 短期处理下放牧强度对土壤真菌群落结构的影响
多年禁牧试验表明放牧通过提高碱性土壤pH和土壤温度增加青藏高原土壤真核生物的多样性与丰富度[16];但也有研究表明放牧强度对真菌群落的影响是通过提高子囊菌门的相对丰度,进而通过竞争排斥的过程间接降低真菌丰富度与多样性[26]。通过对短期放牧不同强度真菌群落的组成和结构进行比较发现,放牧处理均增加了土壤中子囊菌门的相对丰度,但真菌丰富度和多样性均呈现大于对照组的趋势,与之前的研究结果并不完全相同。本研究各处理间的α多样性指数统计学差异不显著,其可能的原因是高寒牧场的土壤性质与微生物群落对短期放牧强度梯度处理具有较强的抵抗力[27];另一方面,由于试验区域较大从而导致土壤微生物空间异质性较高。
本研究结果表明,土壤因子解释了真菌群落变异的39.1%,是真菌群落变化的主要驱动因子,与之前的研究结果相似[14-15]。作为研究区土壤真菌的主要组成部分,放牧处理的子囊菌门相比于对照组均有不同程度的增加,轻度放牧与重度放牧的子囊菌门相对丰度明显增加,与杨文高[28]在牦牛粪便分解试验中牛粪分解增加子囊菌门丰度的结果一致。子囊菌门是牛粪分解过程中的主导类群,其在牛粪分解过程中分泌大量的纤维素与半纤维素分解酶,并利用牛粪中的有机质与营养成分[29],同时放牧使植物与牛粪碎屑等混入土壤中,使土壤中的纤维素含量增加。此外,子囊菌门的底物具有广谱性,通过不稳定碳激活后,可以利用有机碳等天然稳定结构作为底物[30],因而放牧活动过程中家畜排泄的不稳定碳可能进一步促进子囊菌门对有机质的利用。并且子囊菌门与植物多样性呈正相关,放牧强度增加引起的植物多样性增加,从而增加根系分泌物的种类[31],进一步促进子囊菌门对有机质的利用。土壤氮磷循环具有强烈的相互作用,土壤磷可以促进氮的矿化[32],提高土壤的有效氮进而改变微生物群落。土壤微生物相比于植物来说生长速度更快,其遗传结构对磷有更高的需求,因此土壤微生物比植物更容易受到磷的限制[33]。子囊菌门相比于担子菌门的生长速度更快[34],放牧处理土壤中较高的磷含量为其快速繁殖提供了条件。相反,担子菌门的相对丰度在各放牧处理均降低,对环境扰动的抵御能力低于子囊菌门,其可能原因是担子菌门是主要以木质素为底物的寡养性真菌[35],放牧活动使其底物浓度降低。此外,土壤的物理结构决定了真菌的群落结构,担子菌门真菌的菌丝体结构简单,菌丝分枝角较小,菌丝节间长,菌丝体内部孔隙较少;而作为土壤中另一类主要的真菌类群,子囊菌门菌丝体的孔隙率高,对空间的侵占能力更强[36]。本研究中,轻度放牧和重度放牧的土壤表层容重显著低于对照组,土壤孔隙度的增加为子囊菌门提供了更有利的繁殖条件。
3.2 土壤真菌的共现网络及其模块
微生物网络模块的形成主要有两个原因,一是环境因子对微生物的选择使模块类群具有不同于其他模块的生态位,二是模块类群中的微生物具有较强的相互作用[37]。微生物群落实现其功能有两种不同的形式,较高的微生物丰富度造成较大的功能冗余以确保单个功能;或较高的微生物丰富度形成较大的功能多样性,从而同时支持多个功能[38-39]。本研究的真菌共现网络根据真菌类群的相互作用划分为4个主要的模块,由于其组成结构复杂,加上短期放牧对土壤性质影响较小,放牧强度对模块1、2、3的影响不显著;模块4的相对丰度最小且组成分类更丰富,对土壤条件变化敏感,重度放牧的相对丰度显著高于对照组,表明模块丰度越高(模块结构越复杂),抵抗外界干扰的能力越强。Hernandez等[37]得到的微生物群落网络沿胁迫梯度模块化程度降低的结果,表明高胁迫环境的模块结构更少,即模块结构越大对外界胁迫的抵抗能力越强。这是由于网络中各类群间的联系是有限的,模块结构能够维持其内部微生物类群丰度波动的影响不扩散到其他模块[40]。
通过探究各模块的相对丰度与环境因子间的相关关系发现,植物群落特性对真菌模块的相对丰度的影响较小,影响各模块丰度的主要因素仍为土壤性质。与环境因子对真菌群落结构的影响结果相同,土壤全磷和全钾对模块的相对丰度影响更强。尹亚丽等[41]研究表明土壤全钾对真菌群落的影响并不显著;与之不同的是本研究将真菌群落进行类群划分发现,模块1与土壤全钾呈极显著正相关,而其他3个模块与土壤全钾呈负相关。其可能的原因是模块1真菌的主要营养方式为营腐生,其他类群的腐生菌类群丰度降低,Peng等[42]研究几种腐生菌发现,钾离子通过定殖在植物上的真菌促进植物生长。不同于Xu等[43]得到的土壤速效磷与微生物量磷呈负相关的结果,本试验真菌模块对土壤磷的响应大不相同,模块1与土壤全磷显著正相关,模块2与全磷显著负相关,这是因为土壤全磷对真菌的影响是通过与其共生的植物实现的[19]。但由于不同放牧强度间土壤全磷差异不显著,土壤真菌模块1与模块2对不同放牧强度的响应也不显著。本研究的模块1与模块2是构成真菌群落的主要类群,二者相对丰度对环境因子的响应是相反的,造成这一结果的可能原因是同一模块占据相同的生态位,亲缘关系相近的真菌利用相同的底物,功能也趋于一致。放牧活动对土壤容重有直接的影响,以往的研究表明土壤容重可以通过影响土壤孔隙度和土壤温湿度进而改变微生物的群落,使微生物群落与土壤容重呈负相关[44]。土壤容重变小使其孔隙度增大[45],真菌拥有活跃的菌丝,能够随环境的变化与刺激改变其形态[46],土壤容重变小为真菌菌丝的生长提供了空间。本研究仅有模块3的相对丰度与土壤物理性质存在显著相关关系,并且模块3与碱解氮、速效磷等速效养分呈正相关,其可能的原因是该类群的组成单一,对环境因子敏感[47];模块4的真菌相对丰度最小,但其组成分类更丰富,模块3与模块4的组成与结构差异及其对不同放牧强度的响应表明,亲缘关系较远的真菌模块具有更高的模块内关联复杂性,因而对环境变化具有更强的抵抗力,但这与Wagg等[38]得到的结果相反。
4 结论
短期放牧处理可增加土壤真菌群落的多样性与丰富度,但空间异质性与放牧历时较短使得不同放牧强度的真菌群落结构差异不显著。土壤因子是不同放牧强度土壤真菌群落变化的主要驱动因子,放牧处理通过牛粪返还增加土壤中的不稳定碳和改变土壤孔隙两个方面提高子囊菌门的相对丰度,竞争排斥作用使放牧处理的担子菌门相对丰度减少。通过共现网络模块化分析将土壤真菌分为不同的模块类群发现,真菌各物种间的相互作用使真菌群落构成不同的模块类群,但土壤因子仍是真菌相互作用类群变化的主要预测因子。
