复合型固相萃取-离子对反相液相色谱法测定蔬菜中的灭蝇胺残留
2022-10-24王成龙黄小清林泽珊赵金利谭锦萍
王成龙,刘 佳,王 宇,黄小清,林泽珊,赵金利,谭锦萍,黄 松
(广州市食品检验所 广州 511400)
灭蝇胺(Cyromazine)为三嗪类化合物,属低毒类昆虫生长调节剂,常用于抵御菜豆、黄瓜、韭菜等蔬菜上的美洲斑潜蝇和潜叶蝇[1-2]。灭蝇胺在许多国家获准登记并作为农药使用[3]。在豆类蔬菜与瓜果类水果中,欧盟和美国的最大残留限量为0.01 mg/kg,我国明确规定灭蝇胺的最大残留限量不超过0.5 mg/kg。由于种植方式和农药施用不当等原因,使得灭蝇胺的滥用与违规添加现象时有发生[4-6]。国家市场监督管理总局近年均有通报食用农产品豇豆中灭蝇胺残留超标的案例[7]。灭蝇胺具有极性亲水、强内吸性的特点,易通过生物链富集作用,对人体的生殖系统、免疫系统、神经系统、内分泌系统造成损害,而滥用或违规添加可能会引发潜在的食品安全风险[8-10]。
目前,针对灭蝇胺的检测方法主要有液相色谱法[11]、液相色谱-串联质谱法[12-16]、气相色谱-串联质谱法[17]、分子印迹快检技术[18-19]等。液相色谱-串联质谱法一般采用有机溶剂萃取,灭蝇胺极性大,方法的提取效率不高,且检测成本高昂、不易推广;气相色谱-串联质谱法需要对化合物衍生处理,前处理繁琐,测定干扰大;分子印迹技术存在一定的假阳性,在食品监管中存在结果的可靠性风险。依据《食品安全国家标准食品中农药最大残留限量》[20]的最新规定,灭蝇胺的指定判定检验方法为《蔬菜中灭蝇胺残留量的测定高效液相色谱法》NY/T 1725-2009[21],然而,该方法存在净化效果差,回收率偏低的缺点。该方法采用正相色谱柱——氨基柱进行分离,柱键合相氨丙基易脱落,导致柱寿命较短,柱的维护与储存难度大,分析平衡时间长,不适于批量检测。
本研究基于灭蝇胺检测方法,前处理采用复合型固相萃取净化,在目标物的提取与除杂上兼顾非极性保留与离子保留,改善净化效果并增强方法的选择性。色谱分析时,采用以硅胶基质固定相、离子对试剂为流动相的离子对反相液相色谱法,以使灭蝇胺这类强极性化合物有较好的保留,并期望建立起一个快速、高效、特异性好的检测方法以克服当下蔬菜基质灭蝇胺残留定性、定量相对复杂的监管痛点,为后续检测技术标准的整合更新提供技术参考。
1 材料与方法
1.1 样品与试剂
豆角、黄瓜、大白菜、豇豆等具有代表性的蔬菜采买于广州市场,并置于4 ℃冰箱中保存。
灭蝇胺标准品,德国Dr.Ehrenstorfer 公司;盐酸、碳酸氢胺、磷酸、氨水、氯化铵、乙酸锌、亚铁氰化钾、三氯乙酸,广州化学试剂厂;庚烷磺酸钠,美国REGIS 科技公司;三乙胺,上海安谱实验科技公司;乙腈、甲醇、乙酸乙酯、丙酮(均为色谱纯),德国Merck 公司;MCX 复合型阳离子萃取柱、WCX 弱阳离子萃取小柱、HLB 固相萃取柱,美国waters 公司;SCX 强阳离子萃取小柱、C18 固相萃取柱,上海安谱实验科技公司;实验用水为去离子水(经Milli-Q 超纯水机制备)。
1.2 仪器与设备
ACQUITY 高效液相色谱仪、配PDA detector紫外检测器,美国waters 公司;Poroshell C8 色谱柱(4.6 mm×150 mm,2.7 μm),美国Agilent 科技公司;Kintex PFP 色谱柱 (4.6 mm×150 mm,2.6 μm),美国Phenomenex 公司;BEH C18 色谱柱(2.1 mm×100 mm,1.7 μm)、HSS T3 色谱柱(2.1 mm×100 mm,1.8 μm)、BEH Amide (2.1 mm×100 mm,1.7 μm),均为美国waters 公司;UW6200H 型电子天平,日本岛津仪器有限公司;BackManX-30R 型离心机,美国贝克曼库尔特有限公司;MS3-control 涡旋混合器,德国IKA 集团;N-EvAP氮吹仪,美国Organomation 仪器公司;MIlli-Q 纯水机,美国Millipore 公司;2600TH 超声波清洗机、0.22 μm 水相针式滤膜,上海安谱实验科技公司。
1.3 方法
1.3.1 标准溶液的配制 准确称取25 mg 灭蝇胺于小烧杯中,用去离子水溶解并转移至25 mL 规格的容量瓶中,定容至刻度,配成1 mg/mL 灭蝇胺标准储备溶液,于4 ℃冰箱冷藏保存。用去离子水逐级稀释标准储备液,得到0.1,0.2,0.5,1,2 μg/mL 系列纯溶剂标准工作溶液。
1.3.2 样品前处理 称取10 g (精确至0.01 g)蔬菜样品于50 mL 具塞离心管中,加入7 mL 20 mmol/L 碳酸氢胺溶液,以100 kHz 的频率超声提取10 min。分别于离心管中加入1 mL 220 g/L 乙酸锌溶液和1 mL 109 g/L 亚铁氰化钾溶液的组合沉淀剂,涡旋振荡3 min,以8 000 r/min 转速离心3 min。离心后,转移清液于25 mL 容量瓶中后,以7 mL 20 mmol/L 碳酸氢胺溶液重复提取2 次,合并提取液,以碳酸氢铵溶液定容至刻度。分取10 mL 定容液,待净化。
依次用5 mL 甲醇、5 mL 水活化萃取小柱,将待净化液分2 次(5 mL/次)上样至萃取小柱中。随后添加5 mL 0.1 mol/L 盐酸溶液于小柱,再分别用5 mL 去离子水、5 mL 甲醇淋洗,最后以8 mL 5%(体积分数)氨水-甲醇洗脱,收集洗脱液。将洗脱液于40 ℃下氮吹至近干,以1 mL 去离子水复溶,涡旋混匀3 min 后将复溶液过0.22 μm 滤膜,上机测定。
1.3.3 色谱条件 采用C8 色谱柱分离(4.6 mm×150 mm,2.7 μm);紫外检测波长为215 nm;进样体积10 μL;柱温40 ℃;流动相A 为离子对试剂(庚烷磺酸钠溶液: 吸取7 mL 磷酸于200 mL 水中,加入1 g 庚烷磺酸钠,溶解,再加入10 mL 三乙胺,用去离子水稀释至1 000 mL),流动相B 为乙腈,流动相比例VA∶VB=95∶5,等度洗脱;流速1 mL/min;分析时间12 min。灭蝇胺标准出峰图及紫外吸收全光谱见图1。

图1 灭蝇胺标准化合物色谱出峰及其紫外吸收全光谱图Fig.1 The chromatogram and full spectrum of ultraviolet absorption of cyromazine
2 结果与分析
2.1 提取溶剂与提取方式优化结果
灭蝇胺作为极性农药,其pKa=5.2,具有可离子化的官能团氨基(-NH2)与环丙氨基(-C3H6N)[22]。比较碳酸氢胺溶液与前处理常用的甲醇、乙腈等萃取溶剂的提取效果,见图2。
如图2a 所示,灭蝇胺极性强、脂溶性差,采用有机溶剂提取时回收率均较低。而直接用水提取目标物或采用1%三氯乙酸水溶液提取时,回收率均低于60%。由酸式解离方程[23]可知,当采用25 mmol/L 碳酸氢胺溶液(pH=8.5)提取时,溶剂体系的pH 值与灭蝇胺的pKa值相差大于2,目标化合物灭蝇胺呈中性态,官能团未离子化,此时提取溶剂穿透力强,有利于目标物的提取,且碳酸氢铵水溶液还具有比氨水稳定性更好的优点。本研究采用碳酸氢胺溶液作为提取溶剂。
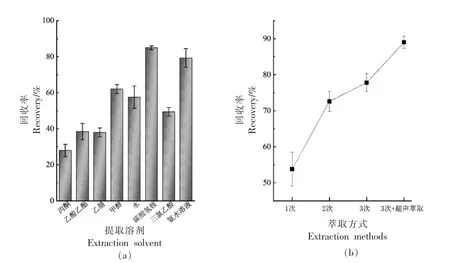
图2 不同萃取溶剂及萃取方式对灭蝇胺提取回收率的影响Fig.2 Effects of different extraction solvents and methods on the recovery of cyromazine
由图2b 可见,采用多次萃取合并提取液方式虽能有效提高灭蝇胺的萃取效率,而反复萃取操作使试验过程繁琐且不利于后续提取液的浓缩。超声辅助萃取通过超声波空化作用增加样品溶剂渗透并加速目标物迁移[24]。采用碳酸氢胺溶液对样品萃取3 次,并在萃取过程中应用超声辅助技术优化,克服了灭蝇胺常规方法前处理提取回收率偏低的问题。本试验采用超声辅助萃取3 次。
2.2 沉淀剂优化结果
测定蔬菜基质中的农药残留,可以通过沉淀植物蛋白、色素等干扰物提高结果的准确性。本研究参照灭蝇胺检测标准[21]、研究文献[25-26]中常用的沉淀剂浓度及用量,比较在前处理中采用不同沉淀剂的沉淀效果,见图3。
图3结果显示:相较于A 组空白对照组,其余组加入沉淀剂,各提取液澄清度均有明显改善,起到沉淀蛋白与部分色素杂质干扰的作用。D 组乙酸锌、亚铁氰化钾溶液组合沉淀剂的沉淀效果最佳,溶液呈澄清透明,且不影响后续测定回收率,故采用乙酸锌、亚铁氰化钾作为前处理方法中的沉淀剂。

图3 基于不同沉淀剂的沉淀效果Fig.3 Effect of various precipitants on vegetable matrices
2.3 固相萃取净化方法优化结果
蔬菜中豇豆基质相对较复杂,含有大量的色素、植物蛋白、皂甙、生物碱等测定干扰物,对样品的除杂与净化有较高要求,同时豇豆也是果蔬中灭蝇胺农药残留的“重灾区”[5],因此选择豇豆做试验样品。分别考察SCX 强阳离子、MCX 混合阳离子、WCX 弱阳离子、C18、HLB 固相萃取小柱(SPE),各SPE 的填料官能团、净化机理、生产厂见表1。其中,在MCX 小柱的使用上,上柱后淋洗过程中,通过盐酸调节pH 值,达到不损失目标化合物的前提下,能分别清除豇豆基质中离子型、极性和非极性干扰物,较好地实现离子交换与非极性保留的优势互补,详见图4。

图4 MCX 固相萃取小柱复合型用法示意图Fig.4 Instruction diagram of MCX composite solidphase extraction column

表1 5 种商品化固相萃取柱填料官能团与净化机理Table 1 Filler functional groups and purification mechanism of five commercial solid phase extraction column
在0.5 mg/kg 加标质量浓度下,各SPE 小柱加标回收率见图5。仅MCX 固相萃取柱满足国标GB/T 27404[27]的要求,因此选择MCX 复合型固相萃取柱为方法净化柱。

图5 不同SPE 小柱净化的回收率比较Fig.5 Comparison of recovery rates of different SPE columns
2.4 色谱柱优化结果
针对灭蝇胺化合物极性强,在反相色谱柱中驻留时间短,出峰较快,难以与极性杂质分开。因此,在使用常规C18 色谱柱(BEH C18)分析之余,考察结合离子对试剂保留的C8 柱(poroshell C8),基于灭蝇胺三嗪环结构特点选取的五氟苯基柱(kintex PFP),针对极性化合物键合三官能团C18 烷基的T3 柱(HSS T3)以及正相色谱柱氨基柱(BEH Amide),先通过优化各柱流动相比例等色谱参数获得灭蝇胺的最佳洗脱条件,再综合比对各色谱柱对灭蝇胺化合物的保留能力 (保留因子k)、出峰峰宽(半峰宽值)、峰形对称性(|对称因子-1|),结果见图6。
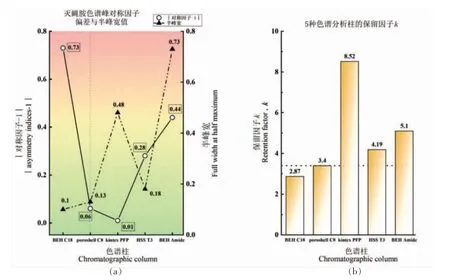
图6 灭蝇胺在各色谱柱上的保留能力、峰宽与峰形对称性Fig.6 Retention capacity,peak width and peak shape symmetry of cyromazine on each chromatographic column
上图中其余色谱柱保留能力均优于常规C18分析柱,唯有结合离子对试剂保留的C8 柱于半峰宽与|对称因子-1|的指标上均能处于低值,即灭蝇胺的出峰达到狭窄、尖锐、对称、不拖尾等要求。本研究选择庚烷磺酸钠为离子对试剂,以C8 色谱柱结合的离子对反相色谱分离体系测定目标化合物灭蝇胺。
2.5 线性范围及检出限
灭蝇胺标准工作液质量浓度分别为0.1,0.2,0.5,1,2 μg/mL,按1.3.3 节所述色谱条件测定,计算峰面积,用最小二乘法进行回归分析。在0.1~2 μg/mL 线性范围的回归方程为y=1.20×105+1.25×103x(y:峰面积响应;x:质量浓度),相关系数R2为0.99871。分别以方法3 倍信噪比(S/N=3)与10 倍信噪比(S/N=10)计算检出限(LOD)为0.012 mg/kg,定量限(LOQ)为0.040 mg/kg。
2.6 加标回收与精密度试验
国标[20]规定灭蝇胺在豇豆中的最大残留限量为0.5 mg/kg。选取豆角、黄瓜、大白菜以及不合格、风险较高的豇豆样品作为4 种代表性蔬菜(样品已用国标方法检测为阴性样品),按1.3.2 节方法做加标试验,其中每种蔬菜选取3 个加标浓度,各浓度测定6 个样品,结果见表2。
如表2所示,各基质平均回收率在77.7%~87.0%之间,相对标准偏差(RSD)在1.6%~3.7%,说明方法稳定可靠,精密度与准确度良好,满足实际测定要求。

表2 加标回收率和精密度试验(n=6)Table 2 Recovery rate and precision experiment (n=6)
2.7 实际样品测定
将建立的方法应用于测定豆角、豇豆等实际检出率较高的样品。从本地农贸市场随机抽取30份该类样品,近半样品中检出灭蝇胺,其含量在0~1.3 mg/kg 范围,存在超标违规现象,继而引发相关的食品安全风险。
3 结论
采用碳酸氢铵溶液结合超声萃取的方式提取灭蝇胺,用复合型固相萃取小柱净化,回收率好,克服了灭蝇胺这类强极性化合物萃取、净化困难的问题。通过优化色谱条件,采用结合离子对试剂保留的C8 色谱柱分离,使灭蝇胺出峰尖锐、对称,方法特异性好,抗干扰强。本方法相较于现行监管判定方法[21],具有灵敏度高、精密度与重现性好的优点,且对复杂基质的除杂效果更为突出。方法的前处理简便、高效,单样测定成本低廉,有利于推广应用,为蔬菜中灭蝇胺残留监管提供技术支持。
