改性处理对绿豆皮膳食纤维结构及功能特性的影响
2022-10-24刘鸿铖樊红秀张闪闪刘婷婷王大为
刘鸿铖,樊红秀,赵 鑫,张闪闪,刘婷婷*,王大为
(1 吉林农业大学食品科学与工程学院 长春 130118 2 农业农村部食用菌加工技术集成科研基地 长春 130118 3 吉林省粮食精深加工与高效利用工程研究中心 长春 130118 4 吉林省粮食精深加工与副产物高效利用技术创新重点实验室 长春 130118)
绿豆 (Vigna radiata (Linn.) Wilczek,mung bean),别名菉豆、青小豆、植豆,为我国传统的一年生豆科草本作物,其种植历史在2000年以上。我国绿豆的种植区域遍布各地,其中以东北地区、淮河及黄河流域的平原地区为主产区,年产量在100 万t 以上[1]。绿豆营养十分丰富,其籽粒中淀粉和蛋白质含量的总和约为80%,此外,还含有多种矿物质、维生素以及脂肪等成分[2-3]。绿豆籽粒种皮的主要成分是膳食纤维(DF),还含有皂苷、黄酮类化合物、蒽醌类及生物碱等多种生物活性物质[4]。
绿豆皮是绿豆的种皮,是绿豆加工企业生产豆芽过程中产出的副产物,目前多用作肥料、饲料贱卖或废渣直接扔掉,未得到充分利用,浪费了这一良好的资源。绿豆皮中膳食纤维的含量在75%以上,是生产高品质膳食纤维的良好材料。膳食纤维由于具有通便排毒,预防心血管疾病,调节血糖等多种生理功能,被世界上公认为七大营养素之一[5]。膳食纤维分为可溶性膳食纤维(SDF)和不溶性膳食纤维(IDF),其中SDF 的生理活性要优于IDF,也被誉为高品质膳食纤维[6]。天然的膳食纤维中SDF 含量较少,IDF 含量过多,导致其口感较差,限制了其在食品配料中的广泛应用。如何采取简便、安全与高效的改性手段,使IDF 尽可能多地转化为SDF,现已成为膳食纤维改性研究的热点[7-8]。
目前,挤出和酶解处理对膳食纤维的改性都有较好的作用,且改性后膳食纤维安全无害,可应用于食品工业。刘婷婷等[9]发现单螺杆挤出改性处理,可以大幅度降低香菇柄纤维的平均相对分子质量和聚合度,持油力、膨胀力等理化指标均明显比改性前增加。张海芳等[10]对比了不同酶法处理马铃薯渣膳食纤维,发现复合酶改性比其它单一酶法改性更能显著提高膳食纤维的理化性质及功能性单糖含量。周爱丽[11]将绿豆皮进行单螺杆挤出改性后,SDF 含量增加到9.27%,提高了绿豆皮膳食纤维孔洞率和表面积,同时未破坏SDF 的分子结构,官能团也未发生变化。朱玉等[12]利用纤维素酶改性小米糠膳食纤维,改性后的膳食纤维胆固醇吸附量比改性前提高了1.40 倍。
目前将挤出处理与酶解改性相结合来改善绿豆皮膳食纤维功能特性的研究还鲜见报道。为提高绿豆皮膳食纤维功能特性,本研究将绿豆皮膳食纤维进行改性处理,以持水力、持油力、膨胀力、阳离子交换能力、吸附葡萄糖能力和吸附胆固醇能力为评价指标,研究改性处理对绿豆皮膳食纤维功能特性的影响。采用扫描电子显微镜、X 射线衍射和傅里叶变换红外光谱法研究改性处理对绿豆皮膳食纤维结构的影响,为提高绿豆皮膳食纤维的利用率和进一步合理应用其营养价值提供理论参考。
1 材料与方法
1.1 材料与仪器
绿豆(一级),购自长春市中东大市场。
碱性蛋白酶(酶活力≥3×103U/mL)、耐高温α-淀粉酶(酶活力≥4×104U/g)、糖化酶(酶活力≥1.6×105U/g)、纤维素酶(酶活力≥1.5×104U/g),诺维信(中国)生物技术有限公司;三羟甲基氨基甲烷,中国医药(集团)上海化学试剂公司;2-(N-吗啉代)乙烷磺酸,中国惠世生化试剂有限公司;葡萄糖,北京化工厂;其它试剂均为市售国产分析纯。
GB1302 电子精密天平,梅特勒-托利多仪器(上海) 有限公司;TU-1901 双光束紫外可见分光光度计,北京普析通用仪器有限公司;GDE-CSF6膳食纤维测定仪,意大利VELP 公司;Nicolet is20傅里叶变换红外光谱仪,美国Thermo Fisher 公司;MiniFlx 600 台式粉末X 射线衍射仪,日本理学株式会社;Phenom Pro 全自动台式扫描电子显微镜,荷兰Phenom-World 公司;JC-60A 型单螺杆挤出试验机,长春市盛达食品工业研究所。
1.2 试验方法
1.2.1 绿豆皮的收集与处理 参照王大为等[13]的方法,并加以修改。挑选表皮无破损、籽粒饱满的绿豆,移入35 ℃的温水中浸泡3 h,除去死豆后将其余绿豆放入25 ℃的豆芽机中进行恒温培养,每30 min 喷淋1 次清水,早晚各换水1 次,连续培养3 d 后即可收集绿豆皮,常温条件下晾干后粉碎至80 目(0.20 mm),置于阴凉干燥处保存备用。
1.2.2 绿豆皮基础组成成分的测定 水分含量测定:直接干燥法,参照《食品安全国家标准 食品中水分的测定》GB 5009.3-2016;脂肪含量测定:索氏抽提法,参照《食品安全国家标准 食品中脂肪的测定》GB 5009.6-2016;灰分含量测定:灼烧重量法,参照《食品安全国家标准 食品中灰分的测定》GB 5009.4-2016;蛋白质含量测定:参照分光光度法,参照《食品安全国家标准 食品中蛋白质的测定》GB 5009.5-2016;总膳食纤维(TDF)、不溶性膳食纤维(IDF)、可溶性膳食纤维(SDF)含量测定:酶重量测定法,参照采用《食品安全国家标准 食品中膳食纤维的测定》GB 5009.88-2014。
1.2.3 绿豆皮的改性处理
1) 挤出改性 取一定量1.2.1 节中的绿豆皮粉,采用单螺杆挤出法进行改性,挤出改性参数为:水分添加量65%(质量分数,下同)、挤出温度160 ℃。物料经过挤出改性后在60 ℃条件下进行热风干燥至恒质量。
2) 酶解改性 取一定量1.3.1 节中的绿豆皮粉,采用纤维素酶解法进行改性,酶解改性参数为:料水比1∶25(g/mL)、纤维素酶添加量0.80%、pH 5.5、反应温度50 ℃、反应时间3.0 h。物料经过酶解改性后在60 ℃条件下进行热风干燥至恒质量。
3) 挤出-酶解复合改性:取一定量1.3.1 节中的绿豆皮粉,采用挤出-酶解复合法进行改性,即将绿豆皮粉加入一定量的水分和纤维素酶进行混合与调配,经润料搅拌均匀后采用单螺杆挤出机进行改性。挤出-酶解复合改性参数为:水分添加量70%、纤维素酶添加量0.30%、pH 5.5、挤出温度140 ℃。料经过挤出-酶解复合改性后在60 ℃条件下进行热风干燥至恒质量。
1.2.4 绿豆皮膳食纤维的提取 参照美国官方分析化学家协会《食物中总的、可溶性及不溶性膳食纤维 酶-重量法》AOAC 991.43 提取绿豆皮膳食纤维。
1.2.5 绿豆皮膳食纤维的结构测定
1.2.5.1 扫描电子显微镜分析 参照Muneer 等[14]的试验方法,取适量干燥粉碎至80 目(0.20 mm)的样品固定于观察台上,离子溅射镀金后放入扫描电子显微镜中,在5.0 kV 加速电压条件下,对样品2 000 倍的表面微观结构进行观察、分析。
1.2.5.2 X 射线衍射测试 衍射条件:采用连续扫描法,管电压:40 kV;管电流:40 mA;靶型:Cu-Kα;扫描范围:5°~45°;扫描速度:2°/min;步长:0.02。
1.2.5.3 傅里叶变换红外光谱分析 通过Nicolet is20 傅里叶变换红外光谱仪测得绿豆皮膳食纤维的傅里叶变换红外光谱曲线[15]。波数范围为4 000~400 cm-1,分辨率为4 cm-1,累计扫描次数为16 次。
1.2.6 绿豆皮膳食纤维功能特性的测定
1.2.6.1 持水力 (持油力) 的分析测定 参照Wang 等[16]的试验方法,并作适当修改。精确称取干燥至恒质量的样品0.20 g 置于15 mL 的离心管中,迅速加入蒸馏水(植物油)10 mL,充分搅拌均匀后用食品保鲜膜密封后在室温条件下放置12 h,然后在室温以4 000 r/min 的转速离心30 min,快速倾析上清液,少量残余的水分(油)用滤纸吸干后,记录样品湿质量,采用公式(1)计算持水力和持油力。
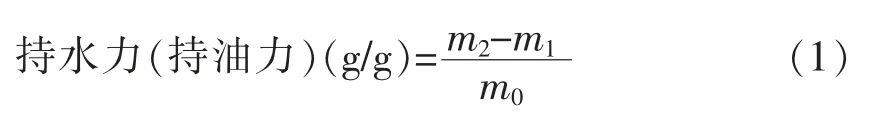
式中:m2——吸水(油)后样品与离心管质量之和,g;m1——离心管质量,g;m0——恒质量样品质量,g。
1.2.6.2 膨胀力的分析测定 参考Chen 等[17]的试验方法,并稍加改动。精确称取干燥至恒质量的样品0.25 g 置于10 mL 量筒中,读取并记录样品自然堆积的体积,然后在室温条件下添加蒸馏水至5 mL 的刻度线,用食品保鲜膜密封,室温静置18 h,读取并记录样品吸水膨胀后的体积,采用公式(2)计算膨胀力。

式中:V1——样品吸水膨胀后测定的体积,mL;V0——恒质量样品自然堆积测定的体积,mL;m——恒质量样品质量,g。
1.2.6.3 阳离子交换能力的分析测定 参照Chau等[18]的试验方法,并适当修改。精确称取干燥至恒质量的样品1.00 g 置于250 mL 的洁净锥形瓶中,添加50 mL 1 mol/L 的HCl 溶液,充分搅拌均匀后用食品保鲜膜密封后在室温条件下放置24 h,以便使样品完全被酸化,然后过滤并用蒸馏水充分洗涤样品至滤液不含Cl-,随后将滤渣移入盛有150 mL 5 g/100 mL 的NaCl 溶液的锥形瓶中,以300 r/min 的转速磁力搅拌30 min,接着滴入2 滴酚酞指示剂,边振荡边用0.05 mol/L 的NaOH 溶液滴定至终点,同时用蒸馏水做空白试验,采用公式(3)计算阳离子交换能力。

式中:V0——滴定空白样所用NaOH 溶液的体积,mL;V1——滴定样品所用NaOH 溶液的体积,mL;m——恒质量样品质量,g;C——滴定所用NaOH 溶液的浓度,mol/L。
1.2.6.4 吸附葡萄糖能力的分析测定 参考Peerajit 等[19]的试验方法,并稍加改动。称取干燥后的样品2.50 g 置于250 mL 的烧杯中,加入150 mL 85%(体积分数) 乙醇溶液,水浴15 min(80℃)后过滤,滤渣经洗涤3 次后在60 ℃条件下进行热风干燥至恒质量。精确称取干燥至恒质量的样品0.50 g 置于250 mL 的洁净锥形瓶中,添加100 mL 浓度为100 mmol/L 的葡萄糖溶液,充分搅拌均匀后移入37 ℃恒温振荡水浴中2,4,6,8,10,12,14,16,18 h,然后迅速取出并以12 000 r/min的转速离心20 min,收取上清液并定容至100 mL。吸取定容后的上清液2 mL,采用DNS 比色法,以葡萄糖标准品绘制葡萄糖标准工作曲线y=0.5344x-0.051 7(R2=0.9973),在540 nm 波长处测定上清液中葡萄糖的浓度,采用公式(4)计算吸附葡萄糖能力。

式中:C1——吸附前葡萄糖溶液中葡萄糖的浓度,mmol/L;C2——吸附后上清液中葡萄糖的浓度,mmol/L;C——葡萄糖溶液的体积,L,本试验为0.1 L;m——恒质量样品质量,g。
1.2.6.5 吸附胆固醇能力的分析测定 参考Zhang等[20]的试验方法,并作适当修改。取若干新鲜鸡蛋并迅速分离出蛋黄,按照1∶9 的体积比加入蒸馏水并充分搅打均匀成蛋黄乳液。精确称取干燥至恒质量的样品0.50 g 置于250 mL 的洁净锥形瓶中,添加30 mL 蛋黄乳液并充分搅拌均匀。然后分别用0.1 mol/L HCl 和0.1 mol/L NaOH 溶液pH 2(模拟胃部环境)和pH 7(模拟小肠内环境)的磷酸盐缓冲液,在37 ℃恒温振荡水浴2,4,6,8,10,12,14,16,24 h 后,然后迅速取出并以4 000 r/min的转速离心20 min,分别吸取上清液1 mL,用邻苯二甲醛法,以胆固醇标准品绘制胆固醇标准工作曲线y=0.0089x-0.0038(R2=0.9915),在550 nm波长处测定上清液中胆固醇的质量浓度ρ1,吸附前蛋黄乳液中胆固醇的质量浓度ρ2,采用公式(5)计算吸附胆固醇能力。

式中:ρ2——吸附前蛋黄乳液中胆固醇的质量浓度,mg/mL;ρ1——吸附后上清液中胆固醇的质量浓度,mg/mL;m——恒质量样品质量,g。
1.3 数据处理与统计分析
使用Origin 8.0、Statistic 软件对试验数据进行处理,测定结果采用“平均值±标准误差”(±s)来表示,图中以标准误差作为误差棒。采用SPSS 22.0 软件 (ANOVA 和Duncan's multiple range test) 对试验数据进行统计分析,P<0.05 时表示为差异显著,P<0.01 时表示为差异极显著。
2 结果与分析
2.1 改性绿豆皮基础组成成分
参照食品安全国家标准测定3 种改性处理方式处理绿豆皮的基础组成成分,绿豆皮改性前后的水分、脂肪、蛋白质、灰分、TDF、IDF 及SDF 含量见表1。绿豆皮经过改性处理后,TDF 的含量没有显著性差异变化,但SDF 含量相较未改性绿豆皮显著性增加(P<0.05),相应的IDF 含量则逐渐减少。由此可见,改性处理可使绿豆皮中部分IDF转化为SDF,尤其是挤出-酶解复合改性处理的效果最明显。这可能是在挤压改性过程中IDF 的一些糖苷键发生断裂,降解成SDF,另一方面纤维素酶可能破坏了绿豆皮细胞壁的结构,从而有利于可溶性成分溶出,这与Li 等[21]得到的结果相似。
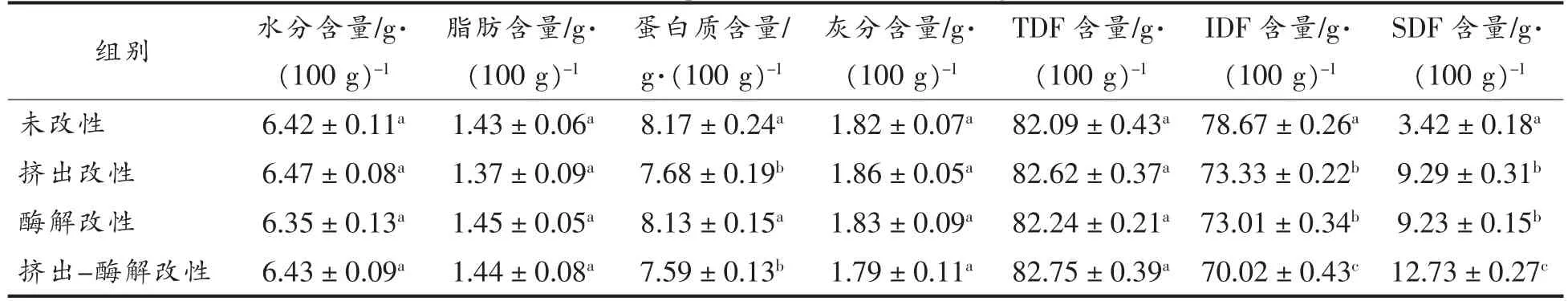
表1 绿豆皮基础组成成分Table 1 Basic components of modified mung bean skin
2.2 表面微观结构观察
图1为2 000 倍的扫描电子显微镜观察改性前后绿豆皮膳食纤维的变化。未改性的绿豆皮膳食纤维表面相对较为光滑,有少量褶皱,结构致密,表面略有粗颗粒附着,可能为绿豆取皮过程中粘连的蛋白质[22](图1a);经挤出改性处理的绿豆皮膳食纤维,表面褶皱较多,结构发生轻微的蓬松,呈蜂窝状的多孔性特征(图1b);经酶解改性处理的绿豆皮膳食纤维,表面变得粗糙,褶皱、孔隙较多,出现不规则颗粒状物质(图1c);经挤出-酶解复合改性处理的绿豆皮膳食纤维,表面变得很粗糙,大量的褶皱和孔隙连成一片,呈多层的疏松网状结构(图1d)。这都为改性处理后的绿豆皮膳食纤维的持水力、持油力和膨胀力的增加提供了依据。樊红秀[23]发现经挤出改性处理后,人参膳食纤维的表面出现多层褶皱与较多大的孔隙,比表面积增大。Yu 等[24]报道了通过不同改性方式处理胡萝卜渣IDF 后,发现其比表面积增大,结构呈疏松多孔状致使持水力增强。上述研究发现均与本文的结果很相似。

图1 改性前后绿豆皮膳食纤维的扫描电子显微镜图Fig.1 Scanning electron microscope (SEM) images of dietary fiber in mung bean skin before and after modification
2.3 X 射线衍射扫描结果
纤维素在固态下有天然纤维素I、人造纤维素II、III、IV 和X 5 种结晶构型。图2为改性前后绿豆皮膳食纤维的X 射线衍射谱图,可以看出在2θ为16.8°和22.4°左右时有明显的结晶衍射峰,且在34.8°处有一个较弱的衍射峰,具有典型的纤维素I 晶体构型[25]。未经改性的绿豆皮膳食纤维的2θ 为16°和22°时出现固有的衍射峰,经挤出改性、酶解改性和挤出-酶解复合改性后的2θ 分别为16.77°,16.83°,16.88°和22.42°,22.38°,22.46°,差异不明显,这说明改性处理并没有使绿豆皮膳食纤维的结晶构型发生显著性改变。改性处理后的绿豆皮膳食纤维结晶区的峰高以及峰面积均有所下降,说明相对结晶度均有不同程度的降低。Jade 7.0 软件拟合后发现,经挤出改性、酶解改性和挤出-酶解复合改性后绿豆皮膳食纤维的相对结晶度分别为37.56%,36.88%,35.17%,相比未改性处理的42.55%,相对结晶度分别降低了11.73%,13.33%,17.34%。这是由于上述3 种改性处理过程中,绿豆皮膳食纤维部分结晶区结构和非结晶区的结构被破坏,分子聚合力降低,使这些结构组分溶出或转化为水溶性成分溶出,一些结晶区转变为非定性区,相对结晶度降低,从而有利于降低绿豆皮膳食纤维的聚合度,导致绿豆皮膳食纤维的持水力、持油力、膨胀力等功能特性的变化[26]。

图2 改性前后绿豆皮膳食纤维的X 射线衍射谱图Fig.2 X-ray diffraction spectra of dietary fiber in mung bean skin before and after modification
2.4 傅里叶变换红外光谱扫描结果
改性处理后的绿豆皮膳食纤维与未改性处理的膳食纤维的傅里叶变换红外光谱分布很相似,但在相应的波数下吸收强度稍有差异。如图3所示,绿豆皮膳食纤维有典型的多糖类特征吸收峰。在3 362 cm-1处出现较强而宽的圆滑吸收峰,这是半纤维素和纤维素的-OH 伸缩振动吸收峰,挤出改性、酶解改性和挤出-酶解复合改性处理后吸收峰强度都稍有减弱,这可能是改性处理过程中纤维素分子间的氢键断裂所致[27],有利于绿豆皮膳食纤维形成多孔结构,持水力增强。2 922 cm-1处的小尖峰是多糖亚甲基和甲基的-CH 拉伸振动吸收峰所致;在1 642 cm-1处的吸收峰是酯类C=O的非对称伸缩振动吸收峰,1 317 cm-1处的吸收峰是木质素强组分的拉伸或弯曲特征吸收峰,经上述3 种改性处理后所对应的特征吸收峰值都稍有减小,这充分说明改性处理后绿豆皮膳食纤维中木质素含量减少,可能是由于经过改性处理后,一些木质素被降解,转化为SDF[28];1 039 cm-1处的较强吸收峰是糖环中醚键(C-O-C)伸缩振动的特征峰[29]。896 cm-1处的弱小尖峰是糖单元中β-型吡喃糖的伸缩振动吸收峰,经过上述3 种改性处理后吸收峰稍有轻微增强,说明改性处理可使糖单元的连接键断裂。610 cm-1处的弱小吸收峰由α-型吡喃糖环的环伸缩振动引起的。上述表明,3 种改性处理绿豆皮膳食纤维的傅里叶变换红外光谱特征吸收峰的强度有着明显的变化,使各纤维素组分得到重新分布,部分IDF 向SDF 转化。

图3 改性前后绿豆皮膳食纤维的傅里叶变换红外光谱图Fig.3 Fourier transform infrared spectra of dietary fiber in mung bean skin before and after modification
2.5 膳食纤维的理化性质分析
如表2所示,改性处理对绿豆皮膳食纤维的持水力、持油力、膨胀力及阳离子交换能力均具有显著性的改善作用,其中,挤出-酶解复合改性处理的效果最好。DF-2、DF-3、DF-4 的持水力分别是DF-1 的1.25,1.28,1.46 倍;DF-2、DF-3、DF-4的持油力分别是DF-1 的1.08,1.11,1.15 倍;DF-2、DF-3、DF-4 的膨胀力分别是DF-1 的1.34,1.22,1.87 倍;DF-2、DF-3、DF-4 的阳离子交换能力分别是DF-1 的4.85,4.32,6.98 倍。这是由于在改性处理过程中,绿豆皮膳食纤维表面出现多层褶皱与较多大的孔隙,比表面积增大,有助于更多的亲水基团和亲油基团暴露出来[30],这使与水和油结合的位点增多,水和油则可以更好地渗透到绿豆皮膳食纤维内部与之结合。另一方面,改性处理也会对绿豆皮膳食纤维中的大分子成分产生一定的破坏作用,使一部分转变为小分子的可溶性成分,有利于持水力、持油力及膨胀力的提高[31]。阳离子交换能力的大小与绿豆皮膳食纤维中的糖醛酸含量密切相关[32],改性处理后的绿豆皮膳食纤维的阳离子交换能力均增强,可能是由于改性处理过程中也把绿豆皮膳食纤维中部分糖醛酸基团暴露出来。这些发现与牛希等[33]采用超声改性处理燕麦膳食纤维得到的结果类似。

表2 膳食纤维的理化性质Table 2 Physiochemical properties of dietary fiber
2.6 吸附葡萄糖能力分析
如图4所示,3 种改性处理绿豆皮膳食纤维对葡萄糖的吸附能力均有显著性提升(P<0.05),且在吸附12 h 后基本达到吸附平衡。在吸附平衡时,DF-2、DF-3、DF-4 对葡萄糖的吸附能力分别由DF-1 的(8.37 ± 0.18) mmol/g 增加到(11.34 ±0.24)mmol/g,(11.06±0.13)mmol/g,(13.89±0.14)mmol/g,即分别是DF-1 的1.35,1.32,1.66 倍。这可能是由于具有黏性的SDF 将葡萄糖进行包裹与束缚,从而把葡萄糖分子截留[34],这与表1中绿豆皮经过改性处理后SDF 含量显著高于改性前SDF 含量结果相符。
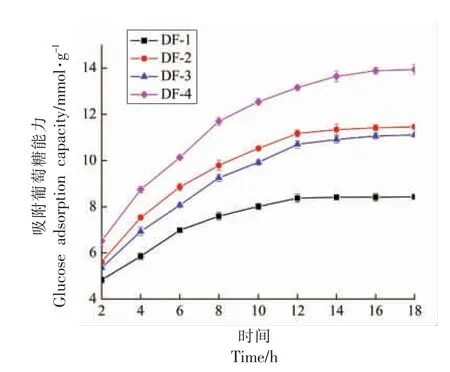
图4 改性前后绿豆皮膳食纤维的吸附葡萄糖能力Fig.4 Glucose adsorption capacity of dietary fiber in mung bean skin before and after modification
2.7 吸附胆固醇能力分析
如图5所示,在pH=2(模拟胃部环境)和pH=7(模拟小肠内环境)条件下,未改性和3 种改性处理后的绿豆皮膳食纤维对胆固醇的吸附能力均随时间的延长呈逐渐上升趋势,在12 h 时基本达到吸附平衡。在相同pH 值条件下,改性处理后绿豆皮膳食纤维的吸附胆固醇能力均显著高于改性前(P<0.05)。在3 种改性处理绿豆皮膳食纤维中,DF-4 对胆固醇的吸附能力最优,在吸附平衡时,DF-4 在pH=2 和pH=7 条件下对胆固醇的吸附能力分别达到(2.38±0.05)mg/(mL·g)和(3.45±0.12)mg/(mL·g)。出现这种现象可能是由于SDF 较IDF分子质量小,极性基团较多,更易于吸附胆固醇[13],这也与表1中绿豆皮经过改性处理后SDF 含量显著高于改性前的结果相一致。Sera 等[35]研究发现,环境pH 值对胆固醇的吸附影响较大,从图5可以看出,同一种绿豆皮膳食纤维,其在pH=7 时的吸附胆固醇能力要优于pH=2。此外,膳食纤维的表面微观结构也在一定程度上影响其吸附胆固醇能力,胆固醇被膳食纤维吸附的过程是一种物理吸附,改性处理后的绿豆皮膳食纤维的表面出现更多褶皱和孔隙,更加有利于吸附胆固醇。

图5 改性前后绿豆皮膳食纤维的吸附胆固醇能力Fig.5 Cholesterol adsorption capacity of dietary fiber in mung bean skin before and after modification
3 结论
本试验研究了改性处理(挤出改性、酶解改性和挤出-酶解复合改性)对绿豆皮膳食纤维结构及功能特性(持水力、持油力、膨胀力、阳离子交换能力、吸附葡萄糖能力、吸附胆固醇能力)的影响。结果表明,3 种改性处理方式均有利于提高绿豆皮中SDF 的含量,其中挤出-酶解复合改性的效果最佳。X 射线衍射、傅里叶变换红外光谱结果显示,各峰强度发生不同程度的改变,这是由于膳食纤维部分结晶区和木质素、纤维素等非结晶区的结构被破坏,一些结晶区转变为非定性区,进而使相对结晶度降低,从而将导致理化特性的变化。绿豆皮膳食纤维经上述3 种改性处理后,表面微观结构均由光滑致密变成不同程度的粗糙与蓬松,出现了多孔性特征或多层褶皱,比表面积大幅度增大。这种改变有利于绿豆皮膳食纤维中更多的亲水基团和亲油基团暴露出来,使持水力、持油力、膨胀力、阳离子交换能力、吸附葡萄糖能力及吸附胆固醇能力等功能特性得到显著改善。通过对3 种改性处理方式进行分析与比较,挤出-酶解复合改性后的绿豆皮膳食纤维,其持水力、持油力、膨胀力、阳离子交换能力等功能特性均为最佳。本研究结果可为改善绿豆皮膳食纤维的结构、功能特性及其广泛应用于功能食品加工中奠定一定的理论基础,对提高绿豆资源的附加值,延长绿豆精深加工产业链具有重要意义及市场前景。
